Tabella T12-1. Classificazione geochimica
degli elementi secondo Goldschmidt.
- Dipartimento di Valorizzazione e Protezione delle Risorse Agroforestali, Università di Torino
- Dipartimento di Scienze Mineralogiche e
Petrologiche, Università di Torino.
- Department of Chemistry and Chemical
Engineering, University of Paisley, Glasgow.
Tesi di Dottorato di Ricerca in Chimica Agraria, XI Ciclo
"Comportamento chimico e mobilità di alcuni metalli pesanti in un'area circostante una fonderia"
Candidato: Luigi Gallini
Ringraziamenti
Rileggendo la tesi che presento mi accorgo che di lei tutto si può osservare eccetto che sia un prodotto finito. Pare piuttosto un percorso intorno ai problemi posti dall'inquinamento del suolo di metalli pesanti. Se qualche risultato conclusivo è stato raggiunto, lo devo alle molte persone che mi hanno aiutato, incoraggiato, indirizzato, ed allietato nell'attività di ricerca bibliografica, di raccolta dei dati e della loro valutazione critica.
Innanzi tutto vorrei ringraziare Aurelio Facchinelli, più un amico che un tutore, sia per quando mi ha, che per quando non mi ha tollerato nei momenti di sconforto. Quindi Marinella Franchini Angela, Emiliano Bruno, Piera Benna, Bruno Alessandria, Vittoria Pischedda, Franco Rolfo ed Elisa Sacchi. Non per ultimi ringrazio Franco Roberto e Fabrizio Negri, del DSMP, e Gianmaria Zuppi del dipartimento di Scienze della Terra.
Ringrazio inoltre il personale del Dipartimento di Valorizzazione e Protezione delle Risorse Agroforestali - Sezione Chimica Agraria. La signora Enza Arduino, innanzi a tutti, che pur essendo andata in pensione ha continuato a prodigarmi delle sue graditissime osservazioni, come peraltro la gentilissima, e colta, signora Elisabetta Barberis. Un ringraziamento particolare va a Eleonora Bonifacio, con la quale ho avuto il piacere di collaborare a lungo e solidalmente, e a Riccardo Scalenghe, Luisella Celi e Maria Martin, con i quali ho divagato a piacere nel magico mondo che orbita attorno alla scienza del suolo. Ringrazio Giovanni Deluca, Bruno Biasiol e il dottor Cignetti senza il cui aiuto non saprei reggere un matraccio misurare un pH o interpretare una misura spettrofotometrica. Un ringraziamento particolare infine al prof. Ermanno Zanini, per avermi gentilmente coinvolto nei suoi interessanti studi sull’erosione. Desidero inoltre ringraziare il numeroso "staff" dell'Università di Paisley, che mi ha generosamente ospitato dieci mesi nonostante le tremende gaffe di cui, come in Italia, sono stato capace. Andrew Hursthouse, che come Aurelio Facchinelli è stato più un amico che un tutore, ma anche Peter, Luise, Bob, Adrian, Jeff, Margaret, Ruth, Steve, Pamela. A Guy Wilthshire rivolgo un ringraziamento particolare per aver messo a punto la determinazione simultanea di As, Bi ed Sb per generazione degli idruri. Un ringraziamento particolarissimo a Mr. David Sterling, per le bellissime digressioni nel campo dello scibile mi ha concesso nel corso delle lunghe misurazioni all'ICP.
Indice
1. Metalli pesanti e suoli .………………………………............................................... 1
1.1. Considerazioni generali ..............................................…………………………............ 1
1.1.1. Osservazioni sul concetto di vulnerabilità associato ai suoli ..........…………………...... 2
1.2 I metalli pesanti ..........................................................……………………………............ 4
1.2.1 Metalli pesanti e suoli agrari .......................................................…………………………... 5
1.2.2. Assimilazione nel vegetale dei metalli pesanti ....................………………………............. 9
1.3. Sorgenti dei metalli pesanti nei suoli ......................………………………….......... 10
1.3.1. Sorgenti naturali, il substrato roccioso ..........................………………………................. 10
1.3.2. Sorgenti antropiche: attività civili, industriali ed agricole .............…………………...... 13
1.3.3. I cicli petrogenici, il comportamento nel suolo e le principali fonti inquinanti di As, Bi, Sb, Pb, Cd, Zn, Cu, Co, Ni e Cr .............………………………........ 16
1.3.3.1. Arsenico (As) .........................................................……………………...................... 18
1.3.3.2. Bismuto (Bi) .......................................................................…………………….......... 26
1.3.3.3. Antimonio (Sb) ..................................................................…………………….......... 38
1.3.3.4. Piombo (Pb) ...............................................................……………………….............. 42
1.3.3.5. Cadmio (Cd) ......................................................................………………………....... 59
1.3.3.6. Zinco (Zn) ...........................................................................………………………...... 68
1.3.3.7. Rame (Cu) .........................................................................…………………………... 70
1.3.3.8. Nichel (Ni) .............................................................................…………………..….... 71
1.3.3.9. Cobalto (Co) .........................................................................……………………….... 71
1.3.3.10. Cromo (Cr) .............................……………………................................................... 72
2. Obiettivi......……………………............................................................................ 73
3. Materiali e metodi .......…………………….................................................... 74
3.1. Inquadramento geologico, pedologico e climatico della Contea di Cliland e del Comune di Villadossola……………………………..…………………………….. 74
3.1.1. La contea di Cliland, Glasgow, Scozia ..…………………………....................................... 74
3.1.2. Il comune di Villadossola, Verbania, Italia ………………………….................................. 77
3.2. Campionamento ...……………………………............................................................. 84
3.3. Preparazione dei campioni ..………………….......................................................
86
3.4. Determinazione dei parametri fisici e chimici dei suoli. ……………. 86
3.5. Trattamento statistico dei dati
....................………………............................... 90
3.6. Determinazione della concentrazione litogenica dell'elemento .......…………………………................................................................... 91
3.7. Determinazione della concentrazione antropica dell'elemento ..............................…………………………............................................ 95
3.8. Determinazione della velocità di lisciviazione degli inquinanti ............................…………………………….................................................. 96
3.8.1. Scelta del modello .................……………………………...................................................... 96
3.8.2. Sviluppo del modello .............…………………………...................................................... 101
3.8.3 Il significato matematico, chimico e fisico del modello a serbatoi e flussi ………….. 104
3.8.3.1. La costante Kn ................………………………........................................................ 104
3.8.3.2. La velocità di lisciviazione Vn ...........……………………....................................... 104
3.8.3.3. Il tempo di semisvuotamento t(1/2)n ...…………………........................................... 105
3.8.3.4 Il tempo di residenza Trn .........……………….................................................. 105
4. Risultati .............……………………................................................................. 106
4.1. I suoli ............................………………………………...................................................... 106
4.1.1. I profili pedologici ..........…………………………............................................................. 106
4.1.2. I fattori di formazione del suolo .............………………………….................................... 115
4.1.3. Influenza della geomorfologia e dell'uso del suolo sulle proprietà chimiche e fisiche dei suoli di Villadossola .……………………….................................. 117
4.1.3.1. Tessitura ................................……………………................................................... 117
4.1.3.2. Sostanza organica ...........……………………......................................................... 123
4.1.3.3. Capacità di scambio cationico ...…………………................................................. 125
4.1.3.4. Il pH ..........................................………………………............................................. 133
4.1.4. Analisi micromorfologiche ...................…………………………..................................... 134
4.1.5 Composizione mineralogica dei suoli di Villadossola .....……………………............... 139
4.2. Metalli pesanti estratti dall’acquaregia .....…………………................... 142
4.2.1. Stima delle concentrazioni litogenica ed antropica ....…………………….................... 142
4.2.2. Stima dell'arricchimento antropico dei metalli pesanti ..……………………................ 145
4.3. La lisciviazione degli inquinanti ....…………………….................................. 148
4.3.1. La velocità di lisciviazione ......................………………………….................................... 148
4.3.2. Il tempo di residenza dell'inquinante nel suolo ....................……………………........... 154
4.3.3. Stima del tempo di autodepurazione dei suoli ........……………........................ 158
4.4. Biodisponibilità di Co, Ni, Cu, Zn, Cd, e Pb nei suoli di Villadossola .....…………………………......................................................................
160
4.6. Distribuzione areale dei metalli pesanti nel comune di Villadossola ....……………………………....................................................................... 165
4.7. Raffronto della concentrazione totale e biodisponibile con i limiti di legge della Regione Piemonte ....……………………....…………………………………... 165
5. Conclusioni .............…………………........................................................... 166
6. Appendici ...........…………………................................................................. 167
6.1. A1. Caratteristiche della stazioni di campionamento ..…………….. 168
6.2. A2.Principali caratteristiche chimiche e fisiche dei suoli ………. 70
6.3. A3. Metalli pesanti in traccia estratti dall'acquaregia .………….... 173
6.4. A4. Elementi maggiori estratti dall'acquaregia ....…..……………...... 176
6.5. A5. Elemento estratto dal reagente di Lakanen .....…………….......... 178
6.6. A6. Principali proprietà chimiche e fisiche dei suoli su cui si è studiata la lisciviazione ......................……………............................ 179
6.7. A7. La costante cinetica del rilascio, la velocità di lisciviazione ed il tempo di residenza ......………………….......................... 181
7. Bibliografia ...................………………….....................................................
183
Per
lungo tempo si è ritenuto che il suolo avesse la capacità di trattenere le
sostanze inquinanti tamponandone gli effetti evidenti entro poco tempo. Si è
quindi sempre prestata più attenzione a quei comparti ambientali, come l’aria o
le risorse idriche superficiali che, invece, reagiscono all’inquinamento
antropico ripercuotendosi sull’ambiente con maggiore immediatezza. La capacità
del suolo d’accumulare le sostanze inquinanti può effettivamente impedire
l’immediata contaminazione d’altri comparti ambientali ma può anche,
determinare un improvviso rilascio degli inquinanti una volta raggiunto il
limite di ritenzione. Per questo motivo si è recentemente rivalutato il
problema dell’inquinamento del suolo, un argomento di cui si dispongono di
relativamente poche informazioni, come può evincersi d’altronde dalle gravi
lacune di cui soffre la legislazione ambientale inerente alla tutela dei suoli.
I potenziali rilasci sono direttamente collegati alla solubilità e alla mobilità dei composti inquinanti poiché, da queste proprietà, dipendono eventuali assorbimenti da parte delle colture agricole, i “flussi” verso le acque superficiali, sotterranee ed oceaniche, e, quindi, pericolose conseguenze per la fauna e la flora del suolo e gli ecosistemi ad esso collegati.
Tra i potenziali inquinanti, particolarmente temuti sono i cosiddetti metalli pesanti i quali, poiché elementi, non sono soggetti ad alcun processo di decomposizione qual è la metabolizzazione microbica che decompone i composti inorganici, e permangono quindi nel suolo fino a che non siano trasportati da qualche meccanismo chimico, fisico o biologico in un altro comparto ambientale.
La presenza di metalli, se in concentrazione superiore a determinate soglie, perturba gli equilibri microbiologici del suolo, condizionandone negativamente la fertilità. I metalli pesanti alterano anche il processo d’assorbimento radicale da parte dei vegetali, con il rischio che una loro eccessiva concentrazione nei suoli adibiti a colture agricole comprometta sia la resa quantitativa del prodotto, che quella qualitativa, determinata dall’ingresso degli inquinanti nella catena alimentare. Anche per i metalli pesanti, come per i composti organici, esiste inoltre il rischio di una discesa verticale attraverso il suolo fino a provocare l’inquinamento delle acque sotterranee.
La determinazione di soglie di nocività per i vari metalli presenti nei suoli rappresenta un problema piuttosto complesso, poiché, oltre a manifestarsi una risposta molto diversa da parte di differenti specie vegetali, anche i comportamenti chimici di tali elementi possono variare molto da suolo a suolo. Questa diversità di comportamenti è presa in considerazione nel concetto di vulnerabilità dei suoli.
Il concetto di vulnerabilità dei suoli è direttamente collegato a quello d’impatto ambientale, inteso come risultante del prodotto di due fattori, la probabilità che occorra un evento indesiderato (rischio) e la gravità delle conseguenze di tal evento (danno). La gravità degli effetti dipende, a sua volta, da due fattori distinti: l’intensità dell’evento stesso (magnitudine) e la risposta dei comparti ambientali coinvolti (vulnerabilità).
Storicamente il concetto di vulnerabilità è stato applicato alle acque sotterranee. Per le acque sotterranee la vulnerabilità è stata quantificata nei termini della minore o maggiore protezione della falda offerta dalla capacità tampone del suolo e della zona non satura soprastanti, ovvero alla capacità del suolo di trattenere gli inquinanti ed impedirne la traslocazione nella falda idrica.
L’applicazione del concetto di vulnerabilità al suolo rappresenta un problema certamente più complesso poiché questo oltre a proteggere le falde idriche dall’inquinamento, svolge molteplici altre funzioni utili (biologiche, ecologiche, agronomiche, ricreative, economiche, geologiche, urbanistiche etc. etc.) ed ognuna di queste funzioni in linea di principio generale viene ad essere compromessa o diminuita in misura diversa da ogni singolo fenomeno o processo che costituisce un rischio per ogni singola funzione utile che esso esercitata. I fattori di vulnerabilità del suolo sono quindi tanto numerosi quante sono le sue funzioni a cui si attribuisce un’utilità.
Ci si può ulteriormente rendere conto della complessità dell’argomento volendo anche solo considerare la vulnerabilità del suolo nei confronti dell’impatto ambientale conseguente l’inquinamento chimico. Un suolo contaminato può, infatti, dare luogo due tipi fondamentali d’impatto ambientale, quello determinato dall’assimilazione dell’inquinante da parte dei vegetali e il conseguente ingresso della sostanza tossica nella catena alimentare, e quello determinato della migrazione della sostanza tossica nelle acque sotterranee. I parametri relativi ai due tipi di vulnerabilità non necessariamente coincidono; infatti, un suolo che, permetta una rapida discesa dell’inquinante verso la falda idrica può essere considerato poco vulnerabile in termini di contaminazione della biomassa vegetale e molto vulnerabile in termini di potenziale contaminazione delle acque.
Le complicazioni aumentano qualora si approfondisca l’analisi della vulnerabilità del suolo nei confronti dei singoli possibili contaminanti e le loro diverse interazioni nei confronti delle diverse fasi che costituiscono il “sistema” suolo; le sostanze organiche presentano, ad esempio, comportamenti differenti da quelli assunti dai composti inorganici ed entrambe le classi di composti al loro interno mostrano ulteriori importanti differenze, quali, per esempio, quelle esistenti fra ioni positivi e negativi o fra molecole polari e non.
Infine, nel caso fosse possibile definire bassa la vulnerabilità di un suolo nei confronti di una determinata sostanza inquinante, sulla base della capacità del suolo a tamponare la sostanza tossica in forme chimiche non assimilate e non lisciviate, va ricordato che tale suolo potrebbe comunque essere considerato vulnerabile rispetto all’improvviso rilascio di inquinante che succede al superamento della capacità tampone o al mutamento delle condizioni pedoambientali (uso del suolo, composizione dell’acqua piovana e di irrigazione, cambiamento climatico etc. etc.).
L’improvviso ed indesiderato rilascio dell’inquinante in forme solubili e biodisponibili è stato definito da Stigliani (1992) come “bomba ad orologeria chimica” (B.O.C.), e, da tempi recenti, è stato ed è oggetto di intensi studi. E’ possibile quindi affermare che un suolo, convenzionalmente definito a bassa vulnerabilità perché dotato della capacità di tamponare elevate quantità di inquinanti in forme non tossiche, presenta un’elevata vulnerabilità in termini di improvvisi ed indesiderati rilasci di sostanze tossiche in forme solubili. L’improvviso rilascio di un metallo pesante si verifica quando:
- l’immissione
del metallo pesante supera la capacità di ritenzione del suolo;
- la capacità d’immagazzinamento di un suolo diminuisce in seguito alla variazione delle condizioni ambientali.
I parametri fondamentali che regalano la capacità del suolo di immagazzinare i metalli pesanti sono: il pH, il potenziale redox, il contenuto di sostanza organica e la capacità di scambio cationico. Le attività umane che possono alterare tali proprietà del suolo sono numerose.
Tra le più rilevanti rientrano le emissioni in atmosfera di sostanze, come ad esempio l’anidride solforosa (SO2) prodotta durante la combustione del carbone e del petrolio, che acidificano le piogge e provocano di conseguenza, l’abbassamento del pH del suolo. I valori del pH del suolo possono subire variazioni anche a causa di pratiche agricole come la calcitazione, che ne provoca l’aumento, o l’uso di fertilizzanti.
Anche il potenziale redox può mutare a seguito dell’intervento antropico, per via del drenaggio di suoli sommersi o a causa dell’irrigazione praticata in quelli aridi al fine di un loro possibile impiego agricolo.
Il contenuto in sostanza organica può essere incrementato dal riciclo dei residui dei raccolti e dall’aggiunta di concime biologico, mentre un inappropriato riciclo della sostanza organica, in pratiche agricole intensive e depauperanti, può causarne un deficit.
La capacità di scambio cationico viene indirettamente alterata dalle attività che modificano il pH, il contenuto in sostanza organica e la salinità della soluzione circolante; quest’ultima, ad esempio, è provocata dall’uso di acque irrigue salmastre, tipico delle aree costiere dove le riserve idriche sotterranee sono sovrasfruttate. La salinizzazione del suolo può causare l’alterazione degli equilibri di scambio ionico (Sequi P., 1989) a favore della solubilizzazione dei metalli pesanti.
Da quanto detto si deduce chiaramente l’importanza che riveste lo studio del comportamento chimico e della mobilità dei metalli pesanti nell’ambito delle indagini ambientali.
- hanno una densità superiore ai 5,0 g/cm3;
- si comportano in genere come cationi;
- presentano una bassa solubilità dei loro idrati;
- hanno una spiccata attitudine a formare complessi;
- hanno una grande affinità per i solfuri, nei quali tendono a concentrarsi;
- hanno diversi stati di ossidazione a seconda delle condizioni di pH ed Eh;
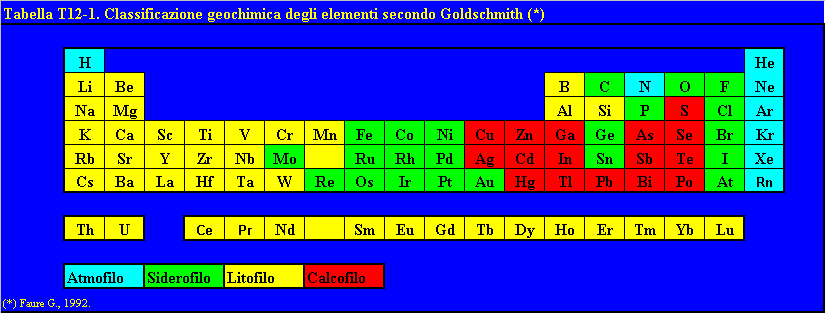
I metalli pesanti, con l’eccezione del Fe e dell’Al, appartengono ai cosiddetti “elementi in traccia”, presenti nei più comuni suoli e rocce della crosta terrestre in concentrazioni inferiori allo 0,1%. Le loro concentrazioni nei suoli, nei sedimenti e nelle rocce sono solitamente di parti per milione o per miliardo. Gli elementi in traccia sono così definiti in contrapposizione agli otto elementi maggiori, O, Si, Al, Fe, Ca, Na, K e Mg che sono presenti nella crosta terrestre in concentrazioni superiori all’1% (Faure, 1992).
Generalmente vengono considerati metalli pesanti l’Ag, il Ba, il Cd, il Co, il Cr, il Mn, il Hg, il Mo, il Ni, il Pb, il Cu, lo Sn, il Tl, il Ti, il V, lo Zn, alcuni metalloidi, con proprietà simili a quelle dei metalli pesanti, quali l’As, l’Sb, il Bi ed il Se (Adriano, 1986. Alloway, 1995. Salomon e Förstner, 1984).
Tra questi, gli elementi che determinano più spesso fenomeni d’inquinamento sono: Cd, Co, Cr, Cu, Mn, Mo, Ni, Pb, Sn e Zn Se (Adriano, 1986. Alloway, 1995. Salomon e Förstner, 1984). Nella sua classificazione geochimica Goldschmidt (Faure, 1992) distingue gli elementi, compresi i metalli pesanti, in (Tabella T12-1):
Tabella T12-1. Classificazione geochimica
degli elementi secondo Goldschmidt.
- siderofili:
aventi affinità per i legami metallici tipici delle leghe;
- calcofili:
aventi spiccata affinità per i legami semimetallici tipici dei solfuri;
- litofili:
caratterizzati dall’affinità per i legami ionici tipici dei silicatici e degli
ossidi;
- atmofili:
aventi bassa affinità per i precedenti legami e pertanto accumulati
nell’atmosfera.
La maggior parte dei metalli pesanti cade nella categoria dei calcofili, fornendo una prima generica indicazione circa il loro comportamento chimico.
Negli ultimi decenni i flussi litosfera-biosfera, litosfera-atmosfera, atmosfera-biosfera relativi a diversi metalli pesanti quali Pb, Hg, Cd, è cresciuto superando abbondantemente quello naturale (Adriano, 1986. Alloway, 1995. Salomon e Förstner, 1984. Mckanzie et al., 1979). In questo contesto si intende con il termine biosfera quel comparto ambientale che contiene il suolo, le acque superficiali e sotterranee, l’atmosfera e la biomassa ed ha solitamente il suolo come “accumulatore” finale dei flussi inquinanti.
Le cause principali dell’alterazione di questi flussi bio-geochimici sono imputabili alla crescente domanda di prodotti contenenti metalli pesanti utilizzati nelle attività industriali e nelle moderne tecniche agrarie ad al conseguente incremento dell’attività mineraria, siderurgica e della produzione e dello stoccaggio dei relativi rifiuti.
Il contenuto totale in metalli
pesanti nei suoli agrari è il risultato degli input provenienti da sorgenti
diverse (figura F121-1) e può essere rappresentato dalla seguente formula:
Mtotale = (Mr + Md + Mf + Mpc + Mro + Mia) – (Ma + Mv + Me + Ml)
dove:
M = metalli pesanti; r = roccia madre; d =
deposizione atmosferica, f =
fertilizzanti; pc = prodotti chimici
di varia natura utilizzati in agricoltura; ro
= rifiuti organici, ia = immissioni
accidentali di varia origine; a =
assimilazione nei raccolti; l =
lisciviazione; v = volatilizzazione;
e = erosione.
La somma (Mp + Ma + Mf + Mac + Mow + Mip)
rappresenta gli apporti complessivi al “sistema” suolo, mentre la somma (Mcr + Mv
+ Me + Ml) costituisce le perdite che
tale “sistema” può subire.
Dal contenuto di elemento nel suolo
(in moli ettaro-1) dividendo per
la velocità delle perdite (moli × anno-1 ha-1) è possibile
stimare il periodo di residenza media (in anni) di ciascun elemento nel suolo.
Si stima per il Cd un tempo di residenza nel suolo compreso tra 75 e 380 anni
e, per elementi più fortemente adsorbiti, quali l’As, il Cu, il Ni, il Pb, il
Se e lo Zn tempi di residenza compresi tra 1000 e 3000 anni. Vengono
considerati degli intervalli di tempo così ampi poiché si è tenuto conto di
tutte le differenti condizioni in cui possono trovarsi i suoli. Il lungo tempo
di residenza indica che un suolo soggetto ad un flusso inquinante tende ad
accumulare la sostanza tossica e raggiungere elevate concentrazioni di
equilibrio con le perdite. Un lungo tempo di residenza indica inoltre che, una
volta cessato il flusso inquinante è necessario un tempo assai lungo affinché la
concentrazione dell’inquinante nel suolo ed i relativi flussi verso i comparti
ambientali ad esso associati (figura F121-2) ritornino ai valori iniziali.
Fino ad oggi si ritiene che alcuni
metalli pesanti non abbiano relazioni dirette con lo sviluppo della biomassa,
anzi si considerano potenzialmente tossici, mentre altri risultano essenziali
per la nutrizione e la crescita di piante ed animali, manifestandosi però
nocivi nel caso in cui le loro concentrazioni superino delle soglie che sono
variabili da elemento ad elemento e da organismo ad organismo (Tabella T121-1).
Per quegli elementi in traccia necessari allo sviluppo degli esseri viventi vengono utilizzati anche i sinonimi “microelementi”, “micronutrienti” ed il termine anglosassone “trace inorganics”. La concentrazione di metallo pesante nel suolo alla quale non si osservano effetti negativi indesiderati,
ovvero “sicura”, è comunque difficile da stabilire in quanto la tossicità può dipendere da fattori che si sommano alla concentrazione dell’elemento.
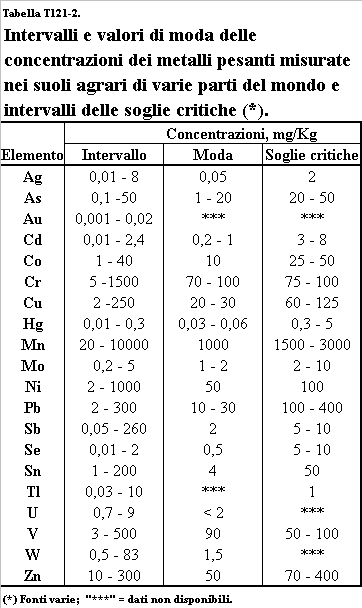
Nella Tabella T121-2 sono riportati gli intervalli entro i quali si situano le concentrazioni dei metalli pesanti osservate nei suoli e, nella seconda colonna, gli intervalli che contengono la maggioranza dei dati osservati. Particolarmente interessante è la terza colonna, in cui sono riportati gli intervalli di valori entro i quali si ritiene possibile la tossicità in qualche forma. Anche questi valori sono dati come intervallo, talvolta anche ampio, in dipendenza della grande incertezza che tuttora permane circa gli effetti di alte concentrazioni. Si può osservare che per molti elementi gli intervalli di concentrazione più frequentemente osservati nei suoli sono prossimi o si sovrappongono alle soglie di concentrazione critiche, giustificando l’attenzione che questi inquinanti ricevono.
Figura F121-1. Bilancio dei guadagni
e delle perdite di metalli pesanti dal suolo.
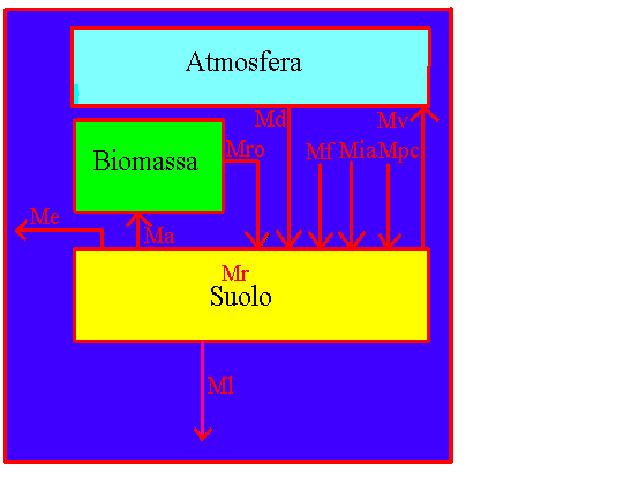
Figura
F121-2. Flusso di materia tra il suolo ed i comparti ambientali ad esso
associati.

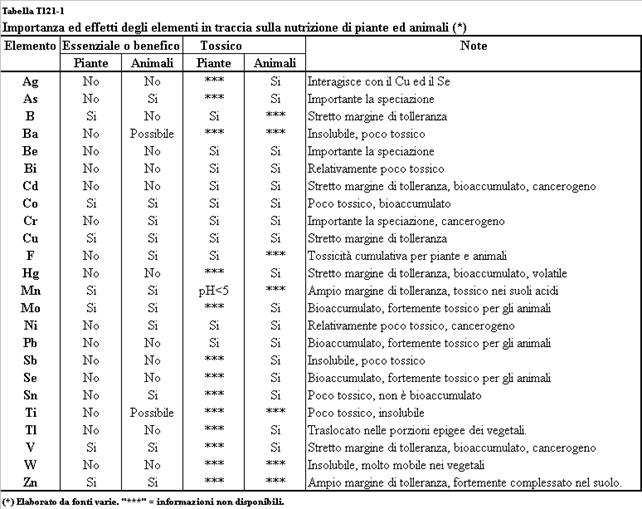
Tabella T121-1. Importanza ed effetti
degli elementi pesanti in traccia sulla nutrizione di piante ed animali
(elaborato da Adriano D.C. (1986) e
Alloway (1995).
Tabella T121-2.
Intervalli e moda delle concentrazioni dei metalli pesanti misurate nei suoli agrari in varie parti del mondo e
soglie di concentrazione critiche.
Il processo di assimilazione degli elementi in traccia da parte dei vegetali può avvenire per trasporto attivo o passivo. Nel processo di trasporto passivo l’elemento viene assimilato senza dispendio di energia da parte del vegetale, mentre nel trasporto attivo l’assimilazione avviene attraverso processi biologici che comportano il consumo di energia. Il trasporto passivo si esplica principalmente attraverso l’assimilazione osmotica. L’assimilazione attiva viene principalmente esplicata dalla pompa protonica ed dal trasporto attraverso specifiche strutture della membrana cellulare dei peli radicali dette cromofori.
Recenti ricerche hanno evidenziato (Gessa C., “Scuola di dottorato in Chimica Agraria” Torino 7-9 maggio 1997, Comunicazione orale) come gli essudati radicali hanno un ruolo di rilievo nell’assimilazione degli elementi di transizione. Gli essudati radicali sono escreti dall’apparato radicale e sono composti da polimeri dell’acido galatturonico. Gli essudati radicali reagiscono con gli ossidi di ferro e di manganese riducendoli. Gli elementi di transizione occlusi negli ossidi di Fe e Mn liberati dal processo riduttivo migrano per osmosi attraverso l’essudato radicale fino a raggiungere la parete cellulare del pelo radicale. Qui possono essere quindi assimilati passivamente per osmosi od attivamente attraverso i gruppi cromofori.
L’assimilabilità dell’elemento è condizionata in modo critico dalle forme in cui esso è presente nel suolo (Page A.L., Miller R.H., e Keney D.R. Eds, 1982). La biodisponibilità degli elementi di transizione decresce progressivamente in dipendenza dell’energia che lega l’elemento alle diverse componenti del suolo. La biodisponibilità è massima per lo ione “libero”, ovvero non complessato e decresce progressivamente dai complessi solubili, alle forme adsorbite aspecificatamente e specificatamente sui colloidi per raggiungere i valori di biodisponibilità minimi nelle forme occluse all’interno dei nei minerali pedogenetici e primari. L’insieme delle forme solubili, adsorbite ed occluse negli ossidi amorfi di Fe Al e Mn prende il nome di frazione labile. Essa assume un particolare rilievo in quanto indagini agronomiche (Page et al., 1982) indicano che la quantità di metallo assimilata dal vegetale è direttamente proporzionale a questa frazione.
Nella modellistica che descrivere i cicli biogeochimici degli elementi mediante modelli a serbatoi e flussi (Lasaga A. C., 1980. Whitfield M, 1981. Lerman A. e Mackenzie F.T., 1975) e nella valutazione dell’impatto sanitario determinato dalla contaminazione dei suoli da metalli pesanti (Sheppard S.C., 1995. Zach R. e Sheppard S. C., 1991) si assume empiricamente che la concentrazione dell’elemento nel vegetale sia direttamente proporzionale alla concentrazione nel suolo, si assume cioè che il rapporto tra la concentrazione dell’elemento nella biomassa vegetale e quella del suolo, detto “fattore di trasferimento suolo-pianta” sia approssimativamente costante (tabella T122-1).
Tabella T122-1.
Fattori di trasferimento suolo-pianta.
Nella tabella sono distinti i fattori di trasferimento suolo-pianta degli organi vegetali vegetativi e quiescenti. Tali fattori sono soltanto indicativi dell’ordine di grandezza del fattore di trasferimento suolo-pianta in quanto, come evidenziato da prove di terreno effettuate in campi minerari (Sheppard S.C. and Eveden W.G., 1990) i fattori di trasferimento risentono fortemente delle forme in cui il metallo è presente nel suolo.
Per una stima più accurata del fattore di trasferimmento suolo-pianta Sillampää M. (1982) impiega due soluzioni estraenti (il reagente di Lakanen e il DTPA). I due reagenti impiegati da Sillampää M. (1982), permettono, tenendo conto della concentrazione volumica del metallo pesante estratto dal suolo e di alcune proprietà chimiche e fisiche del suolo quali il pH, il contenuto in sostanza organica e la CSC, di stimare la concentrazione di Fe, Mn, Cu e Zn nel vegetale con un’incertezza che, pur variando da elemento ad elemento, è in media prossima al 30%.
Un elemento oltre che dall’apparato radicale può essere assimilato dall’apparato fogliare. Considerata l’elevata entità della deposizione al suolo di alcuni metalli pesanti presenti nel particellato atmosferico quali Pb, Cd, Zn, Cu segnalata nelle aree urbane, industrializzate, ed anche molto lontane dai centri industriali (Alloway, 1990. Bertelsen B.O. et al., 1995. Carignan R. e Nriagu J.O., 1985. Shirahata H. et al., 1980) Sheppard S.C. et al. (1992) suggeriscono che nel valutare la biodisponibilità di un elemento e la concentrazione nel vegetale oltre che all’assimilazione radicale si consideri anche l’assorbimento fogliare. Sfortunatamente su questo processo biologico, come osservano gli autori, si dispongono di poche informazioni.
Le possibili sorgenti di contaminazione da metalli pesanti, nell’ambiente in generale e nella pedosfera in particolare, hanno due origini: naturale o antropica. La principale fonte naturale è il substrato geologico mentre tra le sorgenti d’origine antropica le più rilevanti sono dovute alle attività civili ed industriali, responsabili di “input accidentali” legati essenzialmente a sorgenti puntiformi o lineari, ed alle pratiche agricole che rappresentano, invece, “input deliberati”, areali, determinati dalle metodologie utilizzate normalmente in agricoltura.
Nel
corso del processo di alterazione delle rocce il reticolo cristallino dei
minerali primari è distrutto dai processi pedogenetici, ed i metalli pesanti presenti nei reticoli
cristallini primari sono trasferiti nella soluzione circolante del suolo (tabella T131-1).
Una volta raggiunta la soluzione circolante essi possono essere lisciviati
verso la falda idrica od essere occlusi nei reticoli cristallini dei minerali
pedogenetici.
Come regola
generale (Violante P., 1986 a. Violante P., 1989 b.) gli elementi aventi un
elevato rapporto tra carica e raggio ionico, aventi in altri termini un elevato
“potenziale
ionico”, come Fe+3, Al+3, Mn+4, Cr+3
precipitano in forma di idrossidi ed ossidi insolubili. I metalli in traccia a
elevato potenziale ionico come Co, Ni,
Cu, Zn, As, Se, possono vicariare Fe, Al e Mn nei relativi minerali. I metalli
in traccia aventi basso potenziale ionico come Pb e Cd, simili al Ca ed al
K, hanno un raggio ionico troppo elevato
per poter essere ospitati nel reticolo cristallino degli ossidi ed idrossidi di
Fe Al e Mn, ma possono essere adsorbiti in forma scambiabile negli interstrati
di Smectiti, Vermiculiti e Illiti. Gli elementi più facilmente lisciviati e con
i tempi di residenza nel suolo minori sono quelli a basso potenziale ionico
simili ai metalli alcalini ed alcalino terrosi, quali gli elementi delle terre
rare. Tra i processi che trattengono gli elementi in traccia negli orizzonti
superficiali del suolo vi è l’assorbimento nella biomassa vegetale, che imprigiona l’elemento negli
orizzonti organici del suolo attraverso il ciclo suolo-vegetale-suolo. Tra i
processi che favoriscono la lisciviazione dei metalli pesanti vi è la
complessazione con ligandi anionici e la
formazione di specie complesse cariche
negativamente poco trattenute dal complesso di scambio del suolo. Tra i ligandi
che formano i complessi più stabili
con i metalli pesanti vi sono il Cl ed i gruppi carbossilici, fosforici e tiolici della sostanza organica.
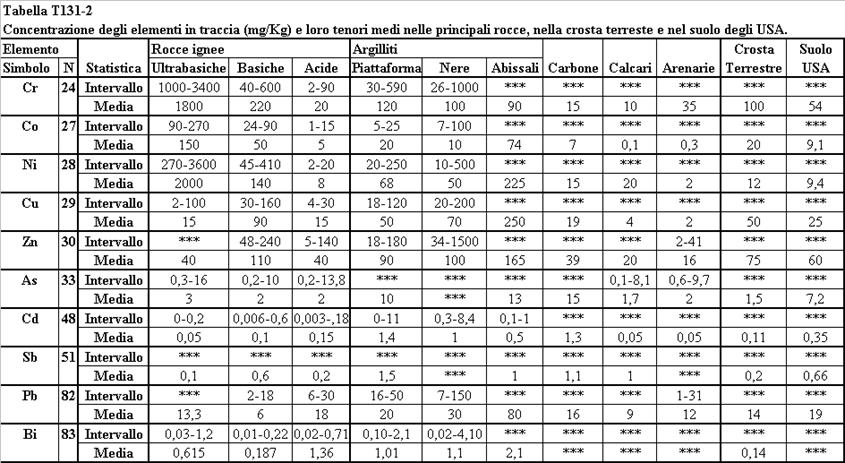
Talvolta nei suoli si osservano elevate concentrazioni di metalli pesanti che possono essere attribuite alla presenza di anomalie geochimiche della roccia madre, come ad esempio capita con il Cr ed il Ni nel caso nei suoli che derivano da un substrato costituito da rocce ultrabasiche e basiche, frequenti in tutti gli orogeni (Negretti G., Sabatino B., 1983). In generale si può osservare che, più un suolo è evoluto, meno è riscontrabile l’influenza della roccia madre nel contenuto di metalli pesanti che presenta il suolo a cui è altrimenti paragonabile (Whedepohl K.H. Ed, 1969).
Per mettere in luce il diverso comportamento dell’elemento nel corso della pedogenesi , di alcuni elementi sono stati riportati i rapporti tra la concentrazione nel suolo rispetto a quella della
roccia madre, detto “fattore di arricchimento nel suolo” (Sposito G., 1989. Vinogradov A.P., 1959., Faure G., 1992). Nella tabella T131-2 sono riportati i fattori di arricchimento osservati nel Nord America negli anni ‘80. Si osserva che alcuni elementi hanno fattori di arricchimento inferiori all’unità, mentre altri come Co e Ni hanno fattori di arricchimento prossimi ad 1. Altri ancora, come Cd, Pb ed As hanno fattori di arricchimento superiori all’unità.
La concentrazione dei metalli pesanti nelle rocce sedimentarie, oltre che dalla composizione mineralogica dei costituenti terrigeni, dipende anche dalla capacità adsorbente del sedimento nonché
dalla concentrazione dell’elemento in traccia presente nelle acque in cui i sedimenti si sono deposti o
sono successivamente venuti a contatto (Bodek I. et al, 1988. Bolth G.H., 1979. Salomons W. e Förstner U., 1984).
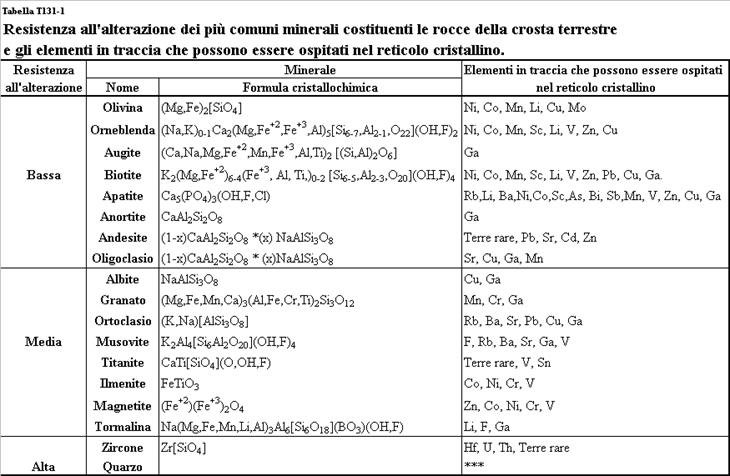
Tabella T131-1. Resistenza all’alterazione dei più comuni costituenti minerali
delle rocce e gli elementi in traccia che possono essere ospitati nel reticolo
cristallino.
 |
La presenza o meno di un determinato metallo pesante in un minerale primario di una roccia ignea o metamorfica dipende dalla sostituzione isomorfa di uno o due degli elementi maggiori con uno o due degli elementi in traccia nella struttura cristallina del minerale. Affinché la sostituzione possa avvenire gli elementi vicarianti devono avere raggio ionico e carica simili. Quando il potenziale ionico dei due elementi differisce meno del 15% i due elementi hanno approssimativamente la stessa probabilità di essere ospitati nel reticolo cristallino. Quando la differenza tra i potenziali ionici supera il 36% la vicarianza tra i due elementi è completa solo alle alte temperature. (Faure G., 1992. Andrew J.E. et al., 1996. Negretti G., Sabatino B., 1983. Wedehpol, 1969). Il potenziale ionico fornisce quindi un’indicazione sulla probabilità che l’elemento in traccia sostituisca l’elemento maggiore e che quindi nel corso della fusione parziale di una roccia, nel coso della cristallizzazione frazionata di un magma o nel processo metamorfico tenda a concentrarsi nella fase liquida piuttosto che in quella solida.
Il comportamento degli elementi in tracce nel processo di differenziazione magmatica e nel processo metamorfico dipende infatti dalla fase mineralogica che cristallizza. Nella formazione di magma basaltico (basico) che ha luogo in corrispondenza delle dorsali medio-oceaniche dalla fusione parziale dai minerali della peridotite del mantello vengono preferenzialmente solubilizzati quelli che hanno un potenziale ionico sensibilmente diverso da quello degli elementi che compongono i minerali principali della peridotite (Plagioclasi, Pirosseni, Spinelli) ovvero quegli elementi che hanno un basso potenziale ionico. Nel corso della cristallizzazione frazionata del magma basaltico vengono rimossi dalla fase fluida quegli elementi che come il Co, il Ni e il Cr vicariano il Fe e il Mg, nei relativi minerali ferro-magnesiaci, caratterizzati da elevata densità e alto punto di fusione. Le rocce acide sono quindi arricchite nei metalli pesanti a basso potenziale ionico quali il Ba ed il Pb che vicariano il Na ed il K nei feldspati alcalini. Distribuzioni più uniformi rispetto al contenuto in silice della roccia hanno il Cu, il Mn, l’As, il B, il Mo ed il Se (Faure G., 1992. Andrew J.E. et al., 1996. Negretti G., Sabatino B., 1983. Wedehpol, 1969). Il comportamento di questi elementi nell’evoluzione magmatica è spiegato da diversi fenomeni, qual è la precipitazione della pirite nei magmi basaltici per il Cu o la capacità di vicariare l’Al, come per l’As. Nella Tabella T131-2 sono indicate le concentrazioni medie di alcuni metalli pesanti nelle rocce più comuni costituenti la crosta terrestre.
1.3.2.
Sorgenti antropiche: attività civili, industriali ed agricole.
Una
delle sorgenti principali di emissioni gassose di metalli pesanti è
rappresentata dai fumi prodotti dal consumo di combustibili per il
riscaldamento (Tabella T132-1);
circa l’84% delle ceneri prodotte dalla combustione dei carboni sono volatili
ed il loro contenuto in elementi in traccia è piuttosto variabile, dipendendo
sia dal tipo di carbone sia dalle condizioni di combustione (Tabella T132-2). Altre importanti fonti di
emissioni in atmosfera contenenti elementi in traccia sono rappresentate dagli
inceneritori di rifiuti e dal traffico veicolare.
Anche
durante il processo estrattivo dei metalli, nonché durante le successive operazioni
di fusione e lavorazione, possono disperdersi nell’ambiente rilevanti
quantitativi di elementi inquinanti attraverso i fumi e le polveri, immessi
dalle ciminiere nell’atmosfera, ed i rifiuti liquidi rappresentati dalle acque
utilizzate durante il ciclo produttivo.
Durante la combustione dei carburanti e dei lubrificanti necessari ai mezzi di trasporto si libera Pb, mentre l’usura dei pneumatici diffonde Zn: in entrambi i casi vi è associata una liberazione di Cd.
Di
particolare interesse e rilevanza è il riutilizzo nell’agricoltura di quei
fanghi, ricchi in sostanze organiche e minerali, prodotti dalla depurazione
delle acque di scarico urbane (reflue), la cui principale limitazione d’uso
dipende dai loro contenuti in metalli pesanti
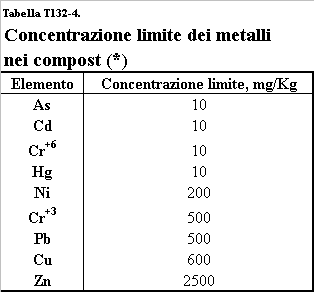
(Adriano D.C., 1986. Alloway B.J., 1997) (tabella T132-3). Anche per i compost, risultato finale di un processo di
trattamento dei rifiuti solidi urbani, la limitazione d’uso, come additivo per
terreni agrari, è rappresentata dalla quantità di metalli pesanti presente (tabella
T132-4). L’applicazione ai suoli di
fanghi di depurazione e/o compost con un eccessivo contenuto in Cd, Cu, Ni, Pb
e Zn in particolare, riduce la resa delle colture o, comunque, peggiora la
qualità dei prodotti; il Cu, il Ni e lo Zn sono risultati i più fitotossici
(vedi anche tabella T121-2). Tra le
fonti d’inquinamento ambientale vanno infine
considerati i possibili rilasci di sostanze tossiche da parte delle
discariche costruite prima dell’entrata in vigore del D.P.R. 915/82.
Per
quanto riguarda gli apporti dovuti alle pratiche agricole, la maggior parte
sono dovuti all’utilizzo dei fertilizzanti sia per il fatto che la concimazione
viene ripetuta stagionalmente sia perché vengono ottenuti dalla lavorazione di
rocce fosfatiche (le Fosforiti) che contengono quantità variabili di As, Cd,
Pb, Bi e Zn (Whedepohl K.H., 1969). Il
Co, il Cu e lo Zn sono contenuti, anche in notevoli quantità, nelle deiezioni
degli animali, che, assimilandone soltanto percentuali molto basse (circa il
5%), fanno sì che le loro feci e le loro urine ne risultano molto arricchite. I
problemi ambientali legati all’impiego delle deiezioni animali nascono, poiché,
soprattutto nelle aziende ad indirizzo zootecnico e cerealicolo-zootecnico,
vengono ridistribuiti su aree limitate o limitatissime per lunghi periodi di
tempo. Stagionali apporti in Cu, Hg, Mn, Pb, As, Sn e Zn sono causati inoltre
dall’uso di pesticidi di varia natura (Adriano D.C., 1986).
Vista
la complessità e la varietà delle possibili fonti d’inquinamento, specie nei
paesi più industrializzati (tabella T132-5),
il monitoraggio e la valutazione delle situazioni a rischio diventa di primaria
importanza per il miglioramento della qualità della vita ed il rispetto
dell’ambiente.
Tabella T132-1. Principali
emissioni di metalli pesanti in atmosfera dell’Europa.
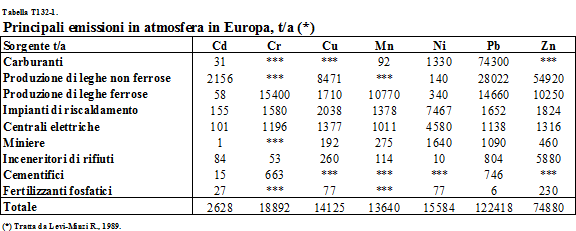
Tabella T132-2. Concentrazioni medie dei metalli pesanti delle ceneri di alcuni
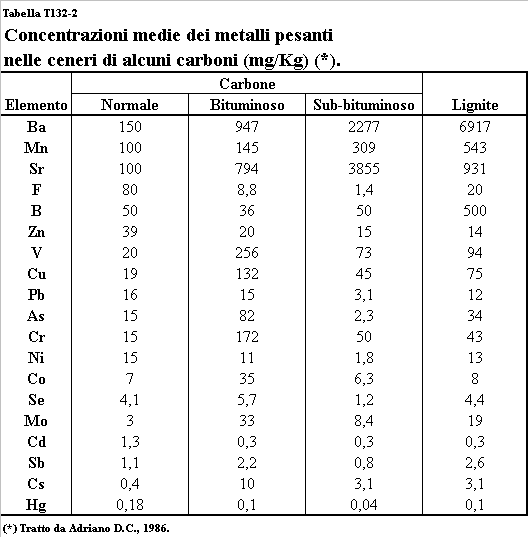
combustibili fossili.
Tabella T132-3. Concentrazione limite dei metalli
pesanti nei fanghi di depurazione utilizzabili come fertilizzanti.

Tabella T132-4. Concentrazione limite dei metalli
pesanti presenti nei compost utilizzabili nella Regione Piemonte.
1.3.3. I cicli petrogenici e il
comportamento nel suolo di As Bi, Sb,
Cr, Pb, Cd, Zn, Cu, Co e Ni.
Il
suolo è definito come “quel sistema: in equilibrio tra litosfera,
biosfera, idrosfera e atmosfera” (Sequi P., 1986). La concentrazione
degli elementi pesenti nei suoli, nelle rocce crostali e negli oceani è infatti
il risultato di equilibri biogeochimici tra l’attività della biosfera e i
processi petrotettonici associati alla tettonica delle placche (figura F133-1). Gli equilibri bio-geo-chimici
riportati nella figura F133-1 sono
descrivibili da modelli a serbatoi e flussi. Questi modelli, a loro volta sono
descritti da sistemi di equazioni differenziali, risolvibili analiticamente nel
caso che tutti i flussi del sistema siano governati da cinetiche del primo
ordine (Lasaga, 1980), oppure numericamente, nel caso siano controllati da
cinetiche di ordine superiore. La sussistenza di un equilibrio tra il suolo, la
litosfera e l’idrosfera è particolarmente evidente se si considera il
coefficiente di ripartizione dell’elemento
tra l’acqua marina e il particellato oceanico è linearmente correlato
sia al coefficiente di ripartizione dell’elemento tra l’acqua marina e la
crosta continentale, che al tempo di residenza dell’elemento nell’oceano
(figura F133-2), (Salomon W. e
Förstner U., 1984). La summenzionata correlazione fornisce indicazioni di
massima sui meccanismi di rimozione dell’elemento dall’acqua oceanica.
Ulteriori informazioni possono essere tratte dalla sua distribuzione attraverso
la colonna d’acqua oceanica (Figura F133-3).
Per alcuni
dei metalli presi in considerazione nel presente lavoro si cercherà nei
successivi paragrafi di riassumerne i cicli petrogenici, il comportamento
dell’elemento nel peodoambiente, nelle acque oceaniche e le principali fonti di inquinamento. In
particolare Lo studio dei cicli petrogenici fornisce indicazioni utili a
comprendere quei meccanismi di quei processi che nel corso delle ere geologiche
hanno mantenuto in equilibrio la concentrazione dei metalli pesanti in traccia
nelle rocce, nei suoli, nelle acque oceaniche e nella biomassa (Salomon W. e
Förstner U., 1984. Whitfield M., 1981. Withfield M. e Turner D.R., 1992).I
meccanismi e i processi che nel corso delle ere geologiche hanno mantenuto in
equilibrio chimico la pedosfera, la biosfera, la litosfera, l’atmosfera e
l’idrosfera, potrebbero permettere infine, mediante un appropriato modello a
serbatoi e flussi, di valutare gli effetti sull’ecosistema planetario
dell’inquinamento del suolo, ed in ultima analisi, di valutare la sostenibilità
delle pratiche agricole ed industriali adottate nei paesi industrializzati
(Lasaga A.C., 1980. Mackenzie et al., 1979. Lerman A., e Mackenzie F.T., 1975.
Lantzy R.J., and Mckenzie F.T., 1979. Schwarzman D.W. e Walk T., 1989).
Nelle
pagine che seguono particolare attenzione è data all’As, Bi, Sb, Cd e Pb. I
primi due elementi si caratterizzano per un marcato carattere semimetallico, e sono presenti nei
suoli in forma di complessi anionici. Il Sb, il Pb ed il Cd hanno carattere metallico marcato, e sono
presenti nella soluzione circolante dei suoli in forma di complessi cationici.
Figura F133-1. Il ciclo biogeochimico di riferimento
(G.E.R.M.) proposto per unificare la modellistica bio-geochimica (*).
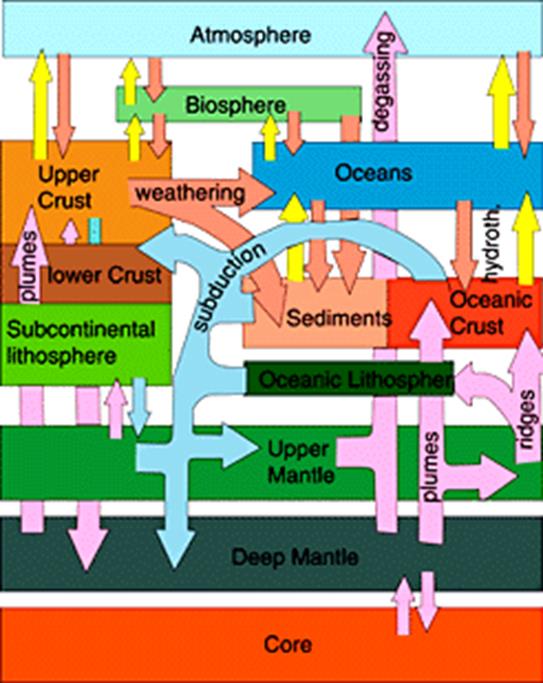
Figura 133-2. Correlazione tra
costante di ripartizione dell’elemento tra l’acqua oceanica e il particellato
oceanico (log Ky) ed il tempo di residenza nell’oceano (ty) (Whitfield M,
1981).
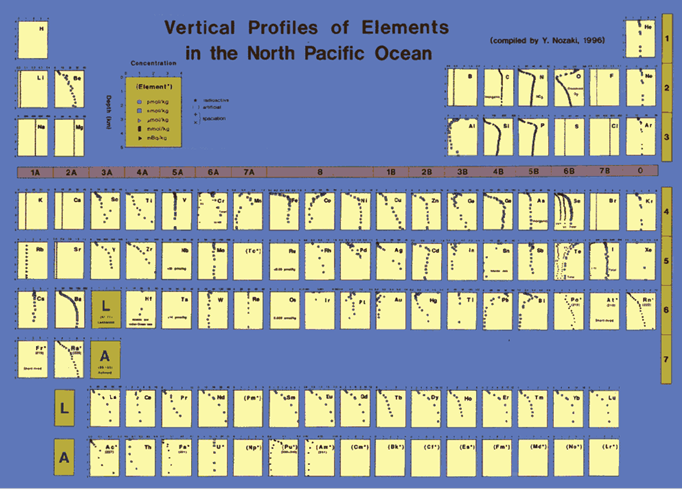
Figura 133-3. Distribuzione degli
elementi nel pacifico settentrionale.
1.3.3.1.
Arsenico (As).
La determinazione dell’As presente in matrici complesse quali rocce suoli e sedimenti è problematica, e le misure effettuate su suoli e rocce standard con metodi differenti concordano a meno di una deviazione standard del 51% (n=5) Pirite (Buaur W.H. e Onishi H., 1972. Nonostante le incertezze analitiche insite nella ardua determinazione dell’As in matrici complesse Buaur W.H. e Onishi H. (1972) compendiano una ricca raccolta di dati petrografici e cristallochimici che permette di tratteggiare il ciclo petrogenico dell’As a seguito riassunto.
La concentrazione media dell’As è di 3 mg/Kg nelle rocce ultrafemiche e di 2 mg/Kg nelle rocce basiche ed ignee, mentre raggiunge concentrazioni medie di 487 mg/l nell’acqua di condensazione delle emissioni fumaroliche (Buaur W.H. e Onishi H., 1972). In corrispondenza delle sorgenti idrotermali delle dorsali medio-oceaniche si osserva la precipitazione di solfuri ricchi in As (Andrews J.E., et al., 1996). Nelle peridotiti che compongono il mantello l’As è contenuto nella magnetite (2,7-41 mg/Kg), nei pirosseni (0,5 mg/Kg), nel plagioclasio (0.14 mg/Kg), nelle Olivine (0,11 mg/Kg) (Buaur W.H. e Onishi H., 1972). In queste rocce, come indicano le regole cristallografiche del Pauling (Faure G., 1992), l’As+3 vicaria l’Al+3 il Fe+3 e il Ti+4. La sostituzione isomorfa dell’Al+3 del Fe+3 e del Ti+4 con l’As+3, a causa dell’elevato raggio ionico dell’As+3, comporta una distorsione dei reticoli cristallini in cui l'As è ospitato e non è quindi favorita. Nel corso della fusione parziale del mantello l’As è quindi espulso dai reticoli cristallini dei minerali primari della peridotite ed è concentrato nella fase fluida. Nel corso della cristallizzazione frazionata del magma basaltico l’As non è efficacemente rimosso dalla precipitazione della Pirite, non essendo questa presente in quantità sufficienti a legare tutto l’As presente nel magma. Essendo l’As+3 uno ione vicariante l’Al+3 il Fe+3 e il Ti+4 esso non è concentrato nei magmi basici più che in quelli acidi. La cristallizzazione dei minerali di Fe, Al e Ti non rimuove completamente l'As dal fuso silicatico. L’As pertanto, si concentra nei fluidi ultracritici espulsi dalla cristallizzazione dei magmi granitici e nei fluidi idrotermali associati ai plutoni e all’attività vulcanica. L’As raggiunge nei fluidi idrotermali di alto grado espulsi dalla cristallizzazione frazionata del magma granitico concentrazioni sufficientemente elevate da precipitare l’Arsenopirite, il principale minerale dell’As, nei filoni tardivi dei corpi granitici e nell’aureola metamorfica ad essi associata (Mastrangelo F., Natale P. e Zucchetti S., 1983). E’ così relativamente frequente osservare negli gneiss che avvolgono i plutoni granitici tenori elevati di As. L’As può essere ospitato in tenori elevati nei solfuri di grado termale più basso, quali Galena, Niccolite, Sfalerite e Pirite. L’intervallo di concentrazioni dell’As che si può osservare in questi solfuri idrotermali è di 50-1000 mg/Kg nella Sfalerite, di 80-1000 mg/Kg, nella Calcopirite, di 80-5000 mg/Kg di 80-20000 mg/Kg e nella Pirite (Buaur W.H. e Onishi H., 1972).Nelle rocce acide, come in quelle basiche, il contenuto di Solfuri ed Arseniati non è sufficiente a sequestrare tutto l’As presente nella roccia e notevoli quantità dell’As presente nel corpo roccioso sono occluse nei silicati primari Pirite (Buaur W.H. e Onishi H., 1972) .
Nell’ambiente pedologico la solubilità dell’As liberato dall’alterazione dei minerali primari essere controllata dalla precipitazioni di fasi insolubili (figura F1331-1). In ambienti riducenti la solubilità dell’As nei suoli è controllata dalla precipitazione dei solfuri, ed in un ristretto campo di valori di Eh, dall’ossido arsenioso As2O3. Nelle condizioni ossidanti le forme prevalenti nei suoli ben aerati sono l’acido arsenico ed arsenioso. L’acido arsenico ed arsenioso a pH superiori a due ed inferiori a 12 sono prevalentemente dissociati nelle forme anioniche H2AsO4-1 ed HAsO4-1. Nonostante l’elevata solubilità dell’As in ambiente ossidante la concentrazione dell’As nella soluzione circolante del suolo è solitamente bassa, 1*10^-2 mg/l (O’Neill, 1997).La concentrazione dell’arseniato e l’arsenito è ovviamente limitata dall’adsorbimento dal complesso di scambio anionico del suolo (Gessa C. e Testini C., 1989. Stumm W., and Morgan J.J., 1996. New Zeland Society of Soil Science, 1980), principalmente determinato da ossidi ed idrossidi di Fe, Al e Mn nonché dalla sostanza organica, dal pH e dalla forza ionica della soluzione circolante (op. cit.). Gli ioni arsenico ed arsenito si combinano con le superfici degli ossidi di ferro attraverso complessi bidentati simili a quelli formati dallo ione fosforico HPO4-2 (O’Neill, 1996). Lo ione fosforico ha una costante di dissociazione simile a quella degli acidi arsenico ed arsinico e pertanto nel pedoambiente compete con l’As nell’adsorbimento sugli ossidi di Fe. Siccome lo ione fosforico ha dimensioni inferiori a quelle dello ione arsenico ed arsinico, forma sulle superfici degli ossidi complessi bidentati più stabili. L’arsenato e l’arsenito adsorbiti sugli ossidi di ferro amorfi eccedono la capacità di scambio anionico del minerale (op. cit.), suggerendo che l’As diffonda all’interno dell’idrossido andando a vicariare il Fe+3 nel solido amorfo. L’adsorbimento dell’As sugli idrossidi di Al cristallini è ben descritto dall’isoterma di (op. cit.) e permette pertanto di escludere la possibilità che l’As migri all’interno di questi minerali cristallini. Studi condotti sui sospensioni di sedimento lacustri indicano che passando da un potenziale ossidativo di +500mV a -200mV e da un pH di 4,0 ad un pH di 7,5 l’As solubile aumenta di 25 volte, in stretta relazione lineare (P<0,01) alla quantità di ferro solubilizzato. Siccome, nel processo di riduzione del sedimento non si ha produzione di composti metilati, l’esperienza prova che alla solubilizzazione degli ossidi di ferro si accompagna il rilascio dell’As adsorbito (op. cit.).
L’acido arsenico ed arsenioso è metabolizzato da funghi e batteri con produzioni di composti metilati volatili e tossici quali l’acido monometilarsonico CH3AsO(OH)2, l’acido dimetilarsinico (CH3)2AsO(OH), l’ossido trimetilarsenico (CH3)3AsO e la trimetilarsina (CH3)2AsH. Le specie metilate prodotte dipendono dalle popolazioni fungine e batteriche (op. cit). Il contenuto di As estraibile in acido fluoridrico attraverso il profilo dei suoli Russi è stato studiato da Vinogradov A.P. (1959). La concentrazione dell’As è risultata compresa nell’intervallo 1 e 10 mg/Kg, con una media di
Figura F1331-1. Diagramma Eh-pH del
sistema As-S-O-H.
(Tratto da Brookis D.G., 1987).
3,6 mg/Kg, valori poco diversi dal valore di 5mg/Kg, risultato della media di 500 suoli raccolti da Vinogradov A.P. (1959) in località provenienti da tutto il mondo. I dati raccolti da Vinogradov A.P. (1959) nella prima metà del secolo indicano che la concentrazione dell’As nel profilo del suolo è principalmente determinata dall’accumulo di sostanza organica e dalla precipitazione degli ossidi di ferro, e che il fattore di arricchimento dell’As nel suolo rispetto alla roccia madre medio nella prima metà del secolo era compreso tra 2,4 e 3,3. I dati raccolti da Schacklette H.T. e Boergen J.B. (1984) indicano che la concentrazione dell’As nel suolo Nord Americano è di 7,2 mg/Kg e il fattore di arricchimento dell’As nel suolo risulta così circa il doppio rispetto a quello misurato da Vinodograv A.P. (1959) nella prima metà del secolo. O’Neill (1997) riporta risultati di recenti rilevi sulla concentrazione di As nei suoli di diversi paesi. La media geometrica della concentrazione dell’As è di 6.7 mg/Kg in Alaska (1988), 40 mg/Kg nel Regno Unito Meridionale (1984), 9,2 mg/Kg in Cina (1991), 2,63 mg/Kg in Polonia (1992), valori per lo più sensibilmente superiori da quelli stimati da Vinogradov (1959).
La biodisponibilità dell’As dipende dalle forme che esso assume nel suolo, ed è maggiore per le specie presenti in soluzione. La frazione di As legata alla sostanza organica è ovviamente facilmente assimilata dai vegetali. Come per il P, la biodisponibilità dell’As è controllata dagli ossidi di ferro (Tamaki S. and Frankenberg W.T. Jr., 1992). Essendo come noto i suoli agrari frequentemente sovrassaturati in P, un efficiente competitore dell’As nell’adsorbimento sugli ossidi di Fe, è ragionevole ritenere che la capacità tampone del suolo nei confronti dell’As sia superata in vaste aree agricole con rischi di rilascio dell’elemento nella catena alimentare e nella falda idrica. Secondo le stime di Baes C. F. et al. (1984) la concentrazione dell’As antropico negli organi vegetativi delle piante si può in prima approssimazione stimare come il 4% dell’As presente nel suolo mentre quella degli organi quiescenti è del 0,6%. Per i suoli Nord Americani, dove il contenuto di As è mediamente di 7,2 mg/Kg, (Shacklette H.T. and Boerngen J.B., 1984) il contenuto nella biomassa vegetale si può stimare di 0,3 mg/Kg. Alla quota assimilata attraverso l’attività radicale, come osservano Sheppard S.C. et al. (1992) la concentrazione dell’As nella biomassa vegetale andrebbe integrata dalla quota determinata dalla deposizione atmosferica sulle piante e dall’assimilazione fogliare, ma, come osservano gli autori, poco è noto sull’assimilazione fogliare degli elementi di transizione in traccia. La quantità di As pedologico inalata può essere stimata se si considera che in media la concentrazione di un elemento nei primi metri a livello del suolo è circa 10^-6 volte la concentrazione dell’elemento nel suolo (Facchinelli et al., 1997). La quantità di As pedologico inalata quotidianamente dalla popolazione Nord Americana può essere stimata quindi in ragione di 0,05 mg/g.
Come indicano Sheppard S.C. et al. (1992) l’ingestione volontaria od involontaria di suolo è una voce importante nel determinare la quantità di elemento in traccia assimilato giornalmente dalla popolazione. Il suolo ingerito accidentalmente dalla popolazione è principalmente costituito dalla frazione argillosa e la quantità ingerita è fortemente correlata all’età (tabella T1331-1). I minerali del suolo hanno scarso effetto nel limitare l’assimilazione dei metalli pesanti (Sheppard S.C., et al., 1994). La quota di As legata al suolo assimilata accidentalmente dalla popolazione dipende quindi criticamente dal contenuto di argilla del suolo, dove l’As, come molti inquinanti, tende a concentrarsi.
La concentrazione dell’As nelle acque dolci continentali è compresa nell’intervallo tra 1*10^-3 e 1*10^-2 mg/Kg e mediamente ha il valore di 2*10^-3 mg/Kg (Tamaki S. and Frankenberg Jr, 1992). Negli ambienti ossidanti tipici delle acque superficiali e delle falde idriche l’As è presente in forme anioniche solubili (figura F1331-1). Le specie prevalenti, in funzione del pH e del Eh, sono H3AsO40, H2AsO4-1, HAsO4-1 ed AsO4-1. La solubilità dell’As nelle acque dolci continentali ben ossigenate è pertanto presumibilmente controllata dalla complesso di scambio anionico e dalla concentrazione delle specie anioniche che competono nell’adsorbimento, le cui principali sono PO4-3, SO4-2, CO3-2 (Schnor J.L., 1996. Stumm W. e Morgan J.J., 1996). Nelle acque oceaniche la concentrazione media dell’As è compresa tra 1,5*10^-3 e 5*10^-3 mg/l ed ha il valore medio di 1,7*10^-3 mg/l (Tamaki S. and Frankenberg W.T. Jr., 1992). La distribuzione della concentrazione dell’As attraverso la colonna d’acqua dell’oceano Pacifico Settentrionale (Nozaky I., 1996) è costante, indicando secondo principi generali dell’oceanografia (Andrews J.E. et al., 1996. Salomon W., and Förstner 1984) che la concentrazione dell’As nell’oceano non è controllata dall’accumulo nel plancton o dalla sedimentazione del particellato oceanico. Nelle rocce sedimentarie la concentrazione dell’As è in ordine decrescente di 15 mg/Kg nel carbone, 10 mg/Kg nelle Argilliti di piattaforma continentale, 1,7 mg/Kg nei carbonati e 13 mg/Kg nelle argille di piana abissale. Nei sedimenti delle piane oceaniche abissali, come è stato evidenziato dall’analisi della varianza (Yuan-Hui Li, 1982) e confermato dall’elevato coefficiente di ripartizione tra solido e liquido dell’As osservato nella cristallizzazione dell’idrossido di ferro a pH 9 (Plotnikow V. I. and Usatova L. P., 1964), l’As è principalmente occluso nelle concrezioni di ossidi ed idrossidi di ferro. La distribuzione dell’As nelle principali rocce sedimentarie suggerisce che il principale meccanismo di rimozione dell’As dalle acque oceaniche sia il seppellimento della sostanza organica e l’occlusione dell’As+3 nei reticoli cristallini degli ossidi di ferro. Dal rapporto della concentrazione dell’As nell’acqua marina e nella crosta continentale il tempo di residenza dell’As nell’oceano può essere stimato in ragione di 10^4 anni (Withfield M., 1975).
Le conoscenze sulla tossicità dell’As sono state riassunte da Tamaki S. and Frankenberg W.T. Jr., (1992). La tossicità dell’As dipende dalla sua forma chimica dell’elemento. L’acido arsenico interagisce con il metabolismo cellulare inibendo la formazione di ATP. L’acido arsenico può inoltre sostituire lo ione fosforico nei fosfoglucidi. L’acido arsinico forma legami stabili con i gruppi reattivi degli enzimi della pelle e dei reni, denaturandoli. L’acido arsinico è più tossico dell’acido arsenico, ed ha un tempo di residenza negli animali maggiore dell’acido arsinico. La dose letale media sui ratti del
Tabella T1331-1. Ingestione
volontaria ed involontaria di suolo.
(Elaborato da Sheppard S.C. et al., 1992).

Figura F1331-2. Ciclo biogeochimico dell’As, dati in 10^8 g.
(Tratto da Mackenzie et al., 1979).
potassio arsenito di 14 mg/Kg e del calcio arsenato di 20 mg/Kg. I composti metilati sono sensibilmente meno tossici. La dose letale media dell’acido dimetilarsinico è di 700-2600 mg/Kg, quella dell’acido metanarsonico è di 700-1800 mg/Kg. L’As è un composto cancerogeno associato ai tumori della pelle.
Le fonti primarie di As sono i giacimenti di Calcopirite e Galena e l’As è un sottoprodotto della raffinazione di Pb e Cu (Alloway D.C., 1986). La forma commerciale grezza dell’As è il triossido arsinico. La produzione mondiale annua di triossido arsinico è stata negli anni ‘70 di circa 47.000 tonnellate. Negli stati Uniti negli stessi anni l’80% dell’As è stato impiegato come defoliante e insetticida a fini agricoli, e l’8% nell’industria ceramica, il rimanente nella concia del cotone (op. cit.). Le principali fonti di inquinamento del suolo dell’As sono quindi lo spargimento dei presidi fitosanitari e le emissioni in atmosfera delle fonderie. Le emissioni in atmosfera di origine antropica sono infatti di 78000 t contro 24000 t di emissioni naturali (Salomons W., Forstner U., 1984). Il ciclo biogeochimico dell’As risulta così fortemente perturbato dalle attività antropiche (figura F1331-2).
1.3.3.2.
Bismuto (Bi).
La tecniche analitiche disponibili per la determinazione del contenuto di Bi nelle matrici complesse quali sono le rocce e i sedimenti, nonostante le ricerche condotte da numerosi chimici analitici (Morrow A., et al. 1997. Whedepol K.H., 1969) sono affette da errori metodologici del superiori al 50%. A causa delle difficoltà poste dalla determinazione della concentrazione del Bi, che richiede attrezzature costose e procedure di analisi complesse, si dispongono di poche ed imprecise misure sulla concentrazione del Bi nelle principali matrici ambientali. Kupcik V. et al.(1978) compendiano un numero numeroso di indagini petrografiche, geochimiche e cristallochimiche sul Bi dalle quali è possibile tratteggiare il ciclo petrogenico a seguito riassunto.
La concentrazione del Bi ha un massimo relativo nelle rocce ultrabasiche (0,62 mg/Kg), un valore minimo in corrispondenza delle rocce basiche (0,19 mg/Kg) ed un valore massimo nelle rocce acide (1,4 mg/Kg) (tabella T131-2). La distribuzione del Bi in funzione del contenuto di silice delle rocce permette di concludere (Negretti G., Di Sabatino B., 1983) che nella fusione parziale delle peridotiti del mantello che genera il magma basaltico il Bi si concentra nel fuso parziale, che di norma costituisce il 2-3% della roccia. Nelle rocce basiche frutto della fusione parziale del mantello il Bi è ospitato nella Pirite, che ha un contenuto medio in Bi di 2 mg/Kg (tabelle T1332-1 e T1332-2), ed nei principali minerali silicatici, dove vicaria il Ca (tabella T1332-4). Nel corso della evoluzione del magma basico il Bi non è efficientemente rimosso dalla cristallizzazione dei Ca-Feldsapati dei Pirosseni, degli Anfiboli e della Pirite, concentrandosi nel fuso silicatico acido. Le concentrazioni massime di Bi vengono così raggiunte nelle rocce acide, nei graniti ed in particolare nei filoni
Tabella T1332-1. Il contenuto di Bi
(mg/Kg) nei solfuri.
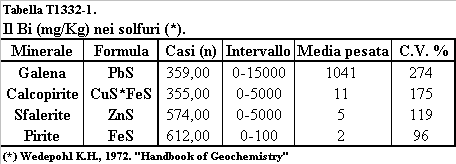
(Elaborato da Kupcik V. et al., 1978).
Tabella T1332-2. Distribuzione di
frequenza del Bi (mg/Kg) nei solfuri.
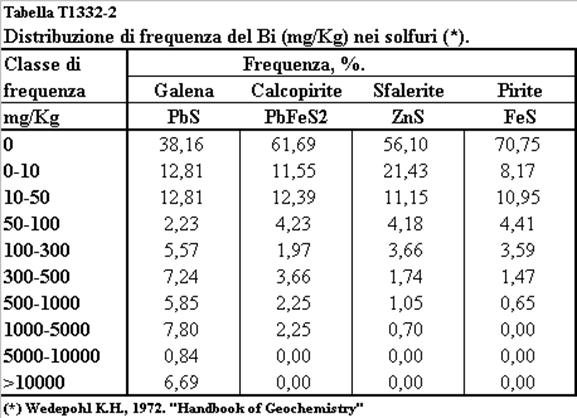
(Elaborato da Kupcik V. et al., 1978).
Tabella T1332-3. La concentrazione
del Bi (mg/Kg) nei minerali delle terre rare. (Elaborato da Kupcik V. et al.,
1978).

Tabella T1332-4. La concentrazione
del Bi (mg/Kg) nei minerali silicatici. (Elaborato da Kupcik V. et al., 1978).

Figura F1332-1. Diagramma Eh-pH del
sistema Bi-O-H-S.
(Tratto
da Brookis D.G., 1988).
pegmatitici, dove il Bi vicaria il Ca nei feldspati potassici, nelle Apatiti e l’Y nei minerali delle terre rare (tabella T1332-3). Nonostante il Bi abbia un potenziale ionico simile al Ca e lo vicari nei minerali silicatici, nelle serie petromagmatiche non si osserva comunque alcuna correlazione tra la concentrazione del Ca e quella del Bi. Il Bi è invece fortemente correlato a quello dell’Y, un elemento delle terre rare (Kupcik V. et al., 1978). Il Bi ha concentrazioni elevate nei solfuri idrotermali, in particolare nella Galena (1041 mg/Kg), nella Calcopirite (11mg/Kg), nella Sfalerite (5 mg/Kg) e nella Pirite (2 mg/Kg) (tabella T1331-2). Il principale solfuro di Bi, la Bismutina Bi2S3 è un minerale relativamente raro, e cristallizza ad un grado idrotermale inferiore a quello caratteristico dei summenzionati solfuri.
Non si dispongono di misure sulla concentrazione del Bi nelle acque dolci superficiali (Salomons W. and Forstner U., 1984. Faure G., 1992. Schnoor J., 1996. Wedepohl K.H., 1969). I dati termodinamici disponibili (figura F1332-1) indicano che nelle condizioni di Eh-pH caratteristiche delle acque superficiali e delle falde idriche la solubilità del Bi è controllata dalla precipitazione dell’ossido Bi2O3 a pH superiore a 6. A pH inferiori il Bi è solubile e la specie prevalente è il Bi6O6+6. Nelle acque oceaniche il Bi ha una concentrazione compresa tra 0,015 e 0,20 g/l, con un valore medio di 0,026+/-37% g/l (Kupcik V. et al., 1978). Le forme chimiche che il Bi assume nell’acqua marina sono BiCl-2, BiOCl+0, BiO+1 (Goldberg E.D., 1965). Attraverso la colonna d’acqua del Pacifico Settentrionale la concentrazione del Bi ha un minimo relativo nei primi 200 m ricchi di vita planctonica, un massimo a cuspide in corrispondenza del limite tra lo strato oceanico superficiale caldo, ossigenato, illuminato, e un decremento esponenziale attraverso lo strato oceanico sottostante freddo, ricco di elementi nutritivi e povero di vita (Yuan-Hui Li, 1982). La distribuzione del Bi attraverso la colonna d’acqua oceanica indica secondo i principi generali dell’oceanografia (Salomon W., and Förstner U., 1984. Andrew J.E., et al., 1996) che questo elemento si comporta come fattore limitante della crescita planctonica nello strato oceanico superficiale, ma che una volta raggiunto il sottostante strato oceanico privo di vita è rapidamente rimesso in soluzione dalla degradazione delle spoglie organiche, adsorbito sul particellato e sedimentato sulle piane abissali. Dal rapporto tra la concentrazione del Bi nella crosta continentale e nell’acqua il tempo di residenza del Bi nell’oceano, a meno della perturbazione antropica degli equilibri naturali, può essere stimato dell’ordine di grandezza di 10^3 anni (Whitfield M., and Turner D.R., 1982. Whitfield M., 1981). Secondo stime più precise (Goldberg E.D., 1965) il tempo di residenza del Bi nell’oceano sarebbe di 45000 anni.
La concentrazione del Bi nelle Argilliti di piattaforma continentale è di circa 1 mg/Kg, un valore di poco superiore a quello della concentrazione media della crosta terrestre, 1,4 mg/Kg. La concentrazione del Bi raggiunge valori nettamente superiori a quelli della crosta terrestre, 2,1 mg/Kg, nelle Argilliti di piana abissale. In questi sedimenti il Bi risulta prevalentemente associato con l’Y presente nei minerali delle terre rare presenti nei noduli di manganese (Yuan-Hui Li, 1982). Tra le concentrazioni massime di Bi si misurano nel carbone e nell'antracite (5,5 mg/Kg), indicando un forte accumulo di questo elemento nella biomassa vegetale dei continenti. Non si dispongono di analisi sul contenuto di Bi nelle rocce carbonatiche (Heinrichs H., Sculz-Dobrick and Wedepohl K.H., 1980. Faure G., 1992. Whedepol K.H., 1969). I dati disponibili sulla distribuzione del Bi nelle rocce sedimentarie indicano che questo elemento è rimosso dall'oceano principalmente dal seppellimento della sostanza organica e dall'intrappolamento nei minerali delle terre rare.
Nelle rocce metamorfiche (tabella T1331-6) la concentrazione del Bi è massima nelle rocce di basso grado metamorfico (Scisti e Filladi) ed è minima nelle rocce metamorfiche di alto grado (gneiss). Nell'alea dei pochi dati disponibili si può concludere che il processo metamorfico ha come effetto la mobilizzazione del Bi. Va osservato che nelle rocce metamorfiche di basso grado il contenuto di Bi aumenta con quello della sostanza organica in perfetto accordo con quanto osservato nelle rocce sedimentarie.
Il Bi può essere presente allo stato metallico in natura solo a pH superiori a 8 ed in ambienti fortemente riducenti eccezionalmente rari nella crosta terrestre (figura F1332-1). In ambiente anossico la sua concentrazione in soluzione è controllata dalla Bismutina Bi2S3, mentre in condizioni ossidanti è limitata dalla precipitazione dell’ossido Bi2O3 a pH superiori a 6. A pH inferiori a 6 la concentrazione del Bi supera le 10^-6 M e non è controllata dalla precipitazioni di minerali insolubili. A pH superiore a 6 la specie chimica prevalente è il Bi6O6+6. Non si conoscono le costanti di stabilità del Bi con i principali ligandi presenti nelle soluzioni circolanti nei suoli o con i principali componenti della sostanza organica così come non si dispongono di informazioni sui meccanismi con cui il Bi è adsorbito dai principali costituenti del suolo (Bolth G. H., 1979. Callahan M. et al., 1979. Yatsimirskii K. B. and Vasil’Ov V.P., 1960. Schwarzen G. and Sillen G. L., 1958). Alloway B.J. (1995), Adriano D.C. (1986), Itamar et. al (1988) ed Alloway B.J. (1997) non riportano informazioni raccolte da indagini condotte sul comportamento del Bi nel suolo. Negli ultimi 10 anni non sono state pubblicate ricerche sul comportamento del Bi nei suoli sulle principali riviste di scienza del suolo: Soil Science Society of America Journal, Canadian Journal of Soil Science e Soil Science. Al riguardo del comportamento del Bi nel suolo si possono avanzare solo ipotesi. Avendo il Bi un potenziale ionico simile al Ca esso potrebbe essere adsorbito in forma idrata nell’interstrato dei minerali argillosi. Secondo le regole cristallochimiche del Pauling, il Bi+3 potrebbe vicariare il Mn nei relativi ossidi. La
struttura elettronica esterna del Bi+3, uguale quella del Pb+2, suggerisce che il Bi possa formare complessi organo-metallici molto stabili. La forte somiglianza tra il comportamento geochimico del Bi con quello delle terre rare, in particolare a quello dell’Y (Kupcik V. et al.,1978), suggerisce che il Bi è rapidamente allontanato dal suolo dai processi pedogenetici.
Tabella T1332-5. La concentrazione del Bi (mg/Kg) nelle rocce sedimentarie.
(Elaborato da Kupcik V. et al., 1978).
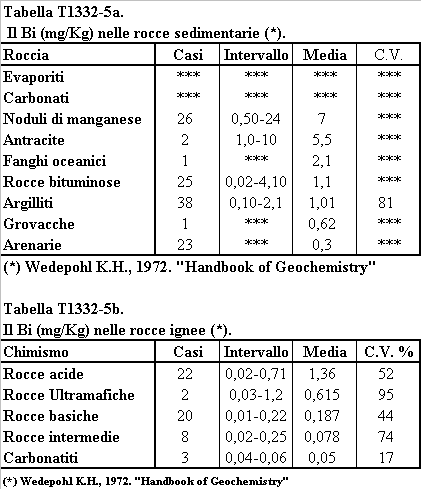
Tabella T1331-6. Concentrazione del Bi (mg/Kg) nelle rocce metamorfiche.
(Elaborato da Kupcik V. et al., 1978).
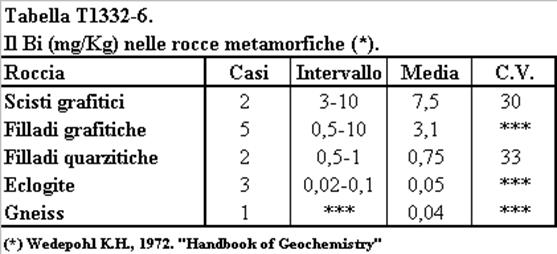
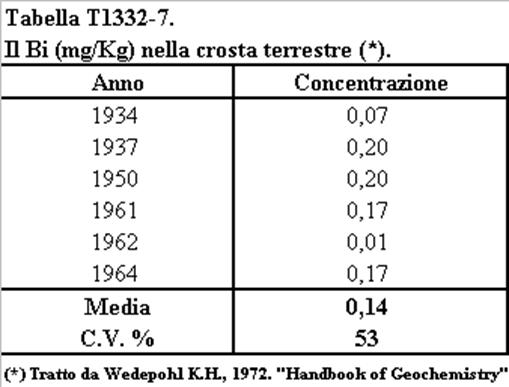
Tabella T1331-7. Stima della concentrazione del Bi (mg/Kg) nella crosta terrestre.
(Elaborato da Kupcik V. et al., 1978).
Poche sono le ricerche
condotte sulla biodisponibilità del Bi. Adriano D.C. (1989), Alloway B.J.
(1995), Coughtrey P.J. et al. (1985), Salomon W. e Förstenr U. (1984) non riportano i risultati di indagini
condotte sulla biodisponibilità del Bi. Baes C. F. et al. (1984) compendiano le
ricerche condotte sulla biodisponibilità del Bi condotte nel corso delle
ricerche sul radioinquinamento. La concentrazione nella biomassa vegetale del
Bi immesso artificialmente nel suolo in forme solubili risulta linearmente
correlato alla concentrazione dell'elemento nel suolo. Negli organi vegetativi
la concentrazione del Bi è mediamente il 3,5% della concentrazione del Bi nel
suolo mentre negli organi quiescenti è solo il 0,6% della concentrazione nel
suolo. Nel computo della concentrazione nella biomassa vegetale del Bi, come
osservano Sheppard S.C. et al. 1992 andrebbe considerata la quota di Bi
assimilata dalle piogge e dal particellato atmosferico su esso deposto.
Non si dispongono di dati sulla tossicità del Bi (Adriano D.C., 1989. Alloway B.J. 1995. Coughtrey P.J. et al., 1985. Salomon W. e Förstenr U., 1984). Nel corso degli ultimi dieci anni le riviste “Environmental Geochemistry and Health” e “Rewiew of Environmental Contamination and Toxicology” non hanno pubblicato ricerche sulla tossicità del Bi. Secondo i criteri proposti da Stumm W. e Morgan J.J., (1996) per valutare la tossicità degli elementi, il Bi dovrebbe essere un elemento potenzialmente tossico sia in quanto capace di formare stabili complessi con i radicali carbossilici dei
composti organici sia in quanto i potenziali di ionizzazione del Bi rientrano nell’intervallo proprio delle reazioni di interesse biochimico. Il Bi non rientra nella lista degli 126 inquinanti a cui l’USEPA da la priorità (Shnorr J. L., 1996).
I minerali del Bi, il cui più diffuso è la Bismutina, sono rari, ed il Bi è estratto principalmente dalla Galena e dalla Calcopirite. Concentrazioni elevate di Bi si trovano anche nei solfuri ed negli Arsenati da cui sono estratti Pb, Cu, Au, Ag e Zn nonché nelle Apatiti. (Wedepohl, 1969). Il Bi è impiegato nella produzione di leghe facilmente fusibili e, in piccole quantità, per indurire i profilati di Pb (Sienko M.J. and Plane R.A., 1980). E’ ragionevole ritenere i suoli possano contenere apprezzabili quantità di Bi provenienti dai fumi emessi dalle fonderie di Pb e Zn e dai fosfati minerali impiegati come fertilizzanti come rilevato per Zn, Cd, Pb ed As (Alloway B.J., 1992., 1984. Adriano D.C., 1986.).
1.3.3.3.
Antimonio (Sb).
Come per As e Bi, le tecniche analitiche non permettono di determinare la concentrazione del Sb con grande accuratezza (Morrow A., et al. 1997. Whedepol K.H., 1969). Il contenuto medio del Sb nelle rocce ignee ultrabasiche, basiche ed acide (tabella T131-2) è di 0,1, 0,6, e 0,2 mg/Kg. Nel corso della fusione parziale delle rocce peridotitiche del mantello esso tende a concentrarsi nel fuso basaltico. Nel corso della cristallizzazione frazionata del magma basaltico esso viene rimosso dalla cristallizzazione dei solfuri, così le che rocce acide hanno una concentrazione di Sb mediamente tre volte inferiore a quella dei basalti. Il Sb che non è rimosso dalla cristallizzazione dei solfuri del magma basaltico può essere ospitato come elemento in traccia nei minerali delle rocce acide, nelle Apatiti o nella Pirite (Wedepohl K. H., 1969). Il Sb vicaria con difficoltà gli elementi maggiori dei minerali delle rocce acide, e nel corso della cristallizzazione frazionata dei plutoni acidi si concentra nei fluidi residuali. Le concentrazioni più elevate di Sb si rinvengono infatti nelle vene idrotermali ad Arsenati o Galena e Calcopirite. I principali minerali del Sb sono l’antimonio nativo Sb, la Stimbnite (Sb2S3), la Kermesite (Sb2S2O), la Senarmonite (Sb2O3), la Jamesonite (2PbS*Sb2O3) e la Boulangerite (5PbS*2Sb2S3) (Wedepohl, 1969). Si dispongono di pochi dati sulla concentrazione del Sb nelle rocce sedimentarie (T131-2), dalle quali risulta che le concentrazioni maggiori di Bi si raggiungono nelle Argilliti di piattaforma continentale. Indagini condotte sulle argille e noduli ferro-manganesiferi delle piane abissali indicano che il Sb, una volta sedimentato sul fondo oceanico, si concentra nelle concrezioni di ossidi di Fe (Yuan-Hui Li, 1982). La concentrazione media del Sb nella crosta continentale è di 0,2 mg/Kg (Shacklette H.T. and Boerngen J.B., 1984. Bowen H. J.. M., 1979).
La concentrazione media del Sb nelle acque continentali dolci è mediamente di 7*10^-5 mg/l mentre nelle acque marine è di 1,5*10^-4 mg/Kg (Faure G., 1992). L’elevato rapporto tra la concentrazione del Sb nelle acque oceaniche e nelle acque dolci superficiali indica che il Sb ha un lungo tempo di residenza nelle acque marine, stimabile in ragione di 10^310^4 anni (Salomons W. and Forstner U., 1984). La concentrazione del Sb attraverso il profilo della colonna d’acqua oceanica (Nozaki Y., 1996) presenta un minimo nei primi 100 m, indice che negli strati superficiali dell’oceano, ricchi di biomassa planctonica, è fortemente bioaccumulato. La concentrazione del Sb attraverso gli strati più profondi si mantiene costante indicando che questo elemento non è efficacemente rimosso dalle acque marine a causa della sedimentazione del particellato sul fondo oceanico o dalla precipitazione di fasi insolubili.
In dipendenza delle condizioni di Eh-pH il Sb può assumere gli stati di ossidazione +3 o +5 (figura F1333-1). La sua solubilità del Sb è controllata dalla precipitazione dei solfuri in ambiente riducenti ed è controllata da ossidi ed idrossidi in ambienti ossidanti. Il Sb può raggiungere concentrazioni in soluzione superiori a 10^-6 M solo a valori di pH inferiori a 2 o superiori a 11 in un ristretto intervallo di potenziali ossidativi alquanto rari nei suoli. Poco si sa sulle forme chimiche che lo Sb può assumere in soluzione (Baes C.F. Jr, and Mesmer R.E., 1976. Callahan M. et al., 1979). I dati disponibili (Bodec I. et al., 1989) indicano nelle soluzioni le specie prevalenti del Sb allo stato di ossidazione +3 sono lo Sb(OH)2+, Sb(OH)30 ed Sb(OH)4-, mentre quelle del stato di ossidazione +5 sono Sb(OH)50 ed Sb(OH)6. Non si conoscono le costanti di stabilità dei complessi che il Sb forma con i principali componenti della materia vivente e della sostanza organica del suolo (Yatsimirskii K. B.
Figura 1333-1. Diagramma Eh-pH del sistema Sb-S-O-H.
(Tratto da Brookis D.G., 1988).
and Vasil’Ov V.P., 1960. Schwarzen G. and Sillen G. L., 1958). Non sono note ricerche condotte sull’adsorbimento del Sb sui minerali del suolo o sulla sostanza organica, ne’ sembra essere stato indagato l’adsorbimento sul complesso di scambio del suolo e dei suoi componenti (Bolth G. H. Ed., 1979. Bodec I. et al., 1988). Secondo Callahan et al. (1979) l’Sb può essere adsorbito sulle argille, coprecipitare ed essere occluso nei sesquiossidi ma avrebbe scarsa reattività con i composti umici. Adriano D.C. (1986) osserva che avendo l’Sb proprietà chimiche e fisiche simili a quelle del P e dell’As esso dovrebbe essere adsorbito dalle superfici degli ossidi Fe, Al e Mn. Infatti da indagini riportate da Adriano D.C. (1986) sulla forma del Sb in sedimenti lacustri inquinati risulta che il 50% del Sb e dell’As sono legati agli ossidi di ferro. Come l’As e l’Hg (Tamaki S. and Frankenberger W.T. Jr, 1992. Bodec I. et al., 1988), l’Sb può essere soggetto a processi biochimici fungini e batterici che determinano la formazione di composti metilici quali Sb(CH3)3 volatili e tossici.
Come riportato da Adriano D.C. (1986) l’Sb trova impiego nella produzione di vernici, ceramiche, vetri e leghe per la confezione di batterie, tubi e laminati. Le principali fonti del Bi sono i giacimenti di Calcopirite, Galena ed Arsenopirite. Elevate concentrazioni di Sb ed As si osservano in
prossimità di fonderie di Cu, Zn e Pb (Adriano D.C., 1986). L’entità del rilascio di Sb nell’ambiente di una fonderia sulla quale sono stati condotti studi di dettaglio (op. cit.) è di 2010^3 Kg/a in atmosfera, e nelle acque superficiali di 210^3 Kg/a in forma solubile e di 1,510^6 Kg/a in forma
insolubile. Berthelsen B.O. et al
(1995) segnalano che attraverso i suoli Norvegesi si osserva un gradiente
negativo nella concentrazione nei suoli del Sb, Pb, Cu e Cd spostandosi dalle
aree industriali settentrionali alle aree agricole meridionali. Nei suoli del
Nord America si osserva un arricchimento del Sb nel suolo rispetto alla roccia
madre di 3,3, che secondo Alloway B.J. (1990) è attribuibile alla deposizione
da atmosfera del particellato prodotto dalle attività industriali.
Adriano D.C. (1986) riporta che sono state condotte poche ricerche sulla assimilabilità del Sb. Bowen H.J.M. (1979) riporta che l’intervallo di concentrazioni del Sb osservato nei vegetali è compreso tra 1*10^-4 e 2*10^-1 mg/Kg. Secondo Baes et al. (1988) la concentrazione del Sb antropico nella biomassa vegetale è efficacemente descritta da una relazione lineare, ovvero dal cosiddetto “fattore di trasferimento”, il rapporto tra la concentrazione dell’elemento estraibile per attacco acido dal suolo e la concentrazione nel vegetale. Il fattore di trasferimento del Sb antropico dal suolo ai
vegetali è mediamente del 20% per gli organi vegetativi e del 3,9% per gli organi quiescenti. Considerato che il contenuto medio del Sb nei suoli del Nord America è di 0,66 mg/Kg si può stimare che la concentrazione media del Sb nei foraggi Nordamericani sia di 0,02 mg/Kg. La concentrazione degli elementi di transizione in traccia presenti nei primi metri a livello di campagna è mediamente un milionesimo della concentrazione dell’elemento nel suolo (Facchinelli et al., 1997), così che l’inalazione del Sb legato ai componenti del suolo può essere stimata in prima approssimazione mediamente di 0,08 mg/g. Il Sb presente nei suoli può essere assimilato dall’organismo umano attraverso l’ingestione volontaria od involontaria di suolo (Sheppard S.C. et al., 1992. Sheppard S.C. et al., 1994. Sheppard S.C., 1995. ). I soggetti geofagi, circa l’1% della popolazione infantile di età inferiore ai 7 anni, possono arrivare ad ingerire 1-5 g di suolo al giorno, ingerendo mediamente 6,6*10^-3 3,3*10^-2 mg/g di Sb. La quantità di suolo ingerita accidentalmente è compresa tra 10-100 mg/g di frazione prevalentemente argillosa. La quantità di Sb assimilata attraverso l’ingestione di suolo dipende così fortemente da quanto l’Sb è concentrato sulle frazione più fine del suolo.
Come osserva Adriano D.C. (1986) non si dispongono di studi sulla tossicità del Sb, osservazione che sembra confermata dalla mancata pubblicazione di informazioni sulla tossicità del Sb sulla prestigiosa rivista “Reviews of Environmental Contamination and Toxicology” edita dalla casa editrice Springer-Verlag. L’Sb è comunque uno dei 126 inquinanti a cui l’USEPA conferisce la priorità di ricerca (Schnor J.L., 1996).
Le tecniche disponibili per l’analisi del contenuto di Pb nelle matrici complesse come rocce sedimenti non presentano difficoltà analitiche (Alloway B.J. Ed., 1990), forniscono risultati precisi e possono essere effettuate con attrezzature poco costose, qual è lo spettrofotometro, disponibili presso la maggior parte dei laboratori chimici. Rispetto ad elementi quali As, Bi, Sb e la cui determinazione presenta difficoltà analitiche particolari, in letteratura sono riportati i risultati di numerosissime ricerche condotte sul comportamento del Pb in una vasta gamma di matrici ambientali (Sahl K., Doe B.R. e Wedepohl K.H., 1978. Davies B.E., 1992. Bodek I., Lyman W. J., Reehl W. F., and Rosenblatt D.H., Eds., 1988. Adriano D.C., 1986. Salomons W., Förstner U., 1984). Sahl K., Doe B.R. e Wedepohl K.H. (1978) compendiano una ricchissima raccolta di dati chimici, cristallochimici e petrografici che permette di delineare il ciclo petrogenico del Pb a seguito riassunto.
Il contenuto di Pb nelle rocce ultrabasiche, basiche ed acide è rispettivamente di 13, 6 e 18 mg/Kg. Questi dati indicano che il Pb, nel corso della fusione parziale del mantello che ha luogo in corrispondenza delle dorsali oceaniche, come tutti gli elementi aventi un basso potenziale ionico, è espulso dai reticoli cristallini dei minerali primari ed è concentrato nel fuso, di norma il 3-4% della massa rocciosa di partenza (Negretti G. e Di Sabatino B., 1983). Sahl K., Doe B.R. e Wedepohl K.H. (1978) non riportano analisi del contenuto di Pb nei minerali che compongono le peridotiti del mantello terrestre. Secondo le regole cristallochimiche del Pauling (Faure G., 1992) si può prevedere che il Pb vicari il K nei reticoli cristallini dei silicati primari. Infatti nelle serie petromagmatiche si osserva una
Tabella T1334-1. Concentrazione media
del Pb (mg/Kg) nelle principali rocce.
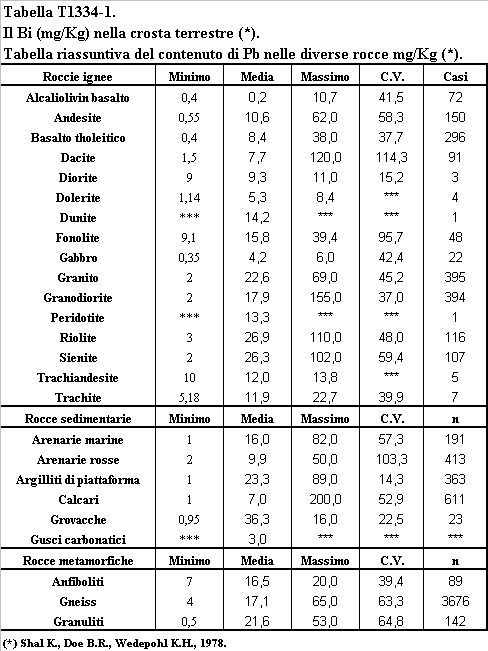
(Tratto da Shan K., Doe B.R., e Wedepohl
K.H., 1978).
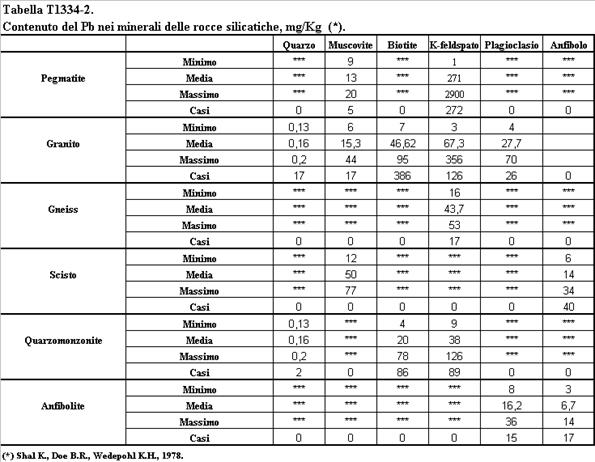
Tabella T1334-2. Concentrazione del Pb
(mg/Kg) nei principali minerali silicatici. (Tratto da Shan K., Doe B.R., e
Wedepohl K.H., 1978).
forte correlazione tra il contenuto di K e Pb delle rocce (Negretti G. e Di Sabatino B., 1983). Nelle peridotiti del mantello esso dovrebbe pertanto essere prevalentemente ospitato nel reticolo cristallino dei Feldspati, nei quali il Pb-Feldspato è parzialmente solubile (Bruno E., and Facchinelli A., 1972). Nel corso del processo di cristallizzazione frazionata del magma basaltico la cristallizzazione dei minerali femici aventi un elevato punto di fusione (Pirosseni, Anfiboli e Ca-Feldspati) è scarsamente efficiente nel rimuovere il Pb dal fuso silicatico (tabella T1334-1) che si arricchisce così in Pb. La cristallizzazione dei solfuri, principalmente della pirite, che ha luogo durante la consolidazione del magma basico, non è in grado di rimuove il Pb dal fuso silicatico in quanto la solubilità della Galena nella Pirite a 700°C è solo del 0,1%. La concentrazione del Pb tende all’aumentare con il grado di evoluzione del magma (tabella T1334-1) ed è minima nei basalti e massima nelle rocce acide.
La concentrazione del Pb nei Graniti, la roccia plutonica più comune nella crosta continentale superiore, è in media di 22 mg/Kg, e ha una distribuzione lognormale (figura F1334-1). La concentrazione del Pb nelle Rioliti è in media lievemente superiore a quella dei Graniti (26,9 mg/Kg) e ha una distribuzione di frequenza normale (F1334-2). Le Rioliti hanno lo stesso contenuto degli elementi maggiori proprio dei Graniti, ma a differenza di questi, hanno avuto modo di risalire attraverso la crosta terrestre e fino a giungere alla superficie terrestre e consolidare in ambiente sub-aereo (Negretti G. e Di Sabatino B., 1983). La differenza nel contenuto di Pb tra graniti e Rioliti indica che nel corso della risalita del leggero magma granitico attraverso la densa crosta terrestre le interazioni chimiche del magma acido con le rocce incassanti e la circolazione dei fluidi idrotermali associati al fenomeno igneo portano ad un arricchimento del Pb nel fuso silicatico. Nei graniti, i solfuri contengono mediamente lo solo lo 0,5% del Pb presente nella roccia. Gran parte del Pb presente nel granito è occluso nel K-Felspato, nella Biotite, nel Ca-Feldspato e nella Muscovite, in un rapporto che è mediamente di 4:3:2:1 (tabella T1334-2). Le concentrazioni maggiori di Pb si osservano nelle Pegmatiti, rocce generate dalla cristallizzazione di fluidi ultracritici espulsi dalla cristallizzazione del magma granitico. In queste rocce la concentrazione del Pb nel K-Felspato è in media di 271 mg/Kg è può raggiungere valori di 2900 mg/Kg (T1334-2). La concentrazione del Pb nei fluidi idrotermali raggiunge concentrazioni sufficientemente elevate per determinare la cristallizzazione della Galena (PbS), il principale minerale del Pb, nell’idrotermalismo di medio grado. Nella Galena sono di norma ospitate ingenti quantità di Shapbachite o Matildite (rispettivamente le forme e di AgBiS), minerale con il quale sopra ai 400°C la Galena ha una miscibilità allo stato solido completa. Nelle emissioni fumaroliche associate all’attività vulcanica l’emissione di Pb è controllata dal composto alogenato più volatile, il PbCl2.
La concentrazione del Pb nelle acque piovane dell’emisfero australe riportata da Sahl K., Doe B.R. e Wedepohl K.H. (1978) è di 0,72+/-20% g/l. Le concentrazioni di Pb più basse (0,162,6 g/l) si osservano in Groenlandia, in prossimità del Circolo Polare Artico, mentre i valori più alti (6,229,2g/l) si osservano nel Michigan, un’area fortemente industrializzata. Sahl K., Doe B.R. e Wedepohl K.H. (1978) compendiano una raccolta di 6562 analisi del contenuto di Pb in campioni di acque dolci continentali provenienti da tutto il mondo, la cui concentrazione media è di 1,78 g/l. La concentrazioni del Pb nelle acque del Nord America risulta sensibilmente più alta, compresa tra 1 e 55 g/l e con un valore medio di 2,51 g/l (n=1233). Sahl K., Doe B.R. e Wedepohl K.H. (1978) stimano la concentrazione del Pb nelle acque oceaniche incontaminate di 2*10-3 g/l. I valori di Pb osservati nelle acque superficiali prossime alle coste continentali sono assai superiori, compresi tra 0,08 e 0,36 g/l ed hanno un valore medio di 0,19 g/l. L’incremento della concentrazione del Pb nelle acque marine è rimasta registrata nell’esoscheletro dei coralli (Andrew J.E., et al., 1996). La concentrazione del Pb nell’esoscheletro dei coralli si mantiene su valori costanti di 10 nM di Pb per ogni M di Ca fino agli anni ‘30. Successivamente si osserva un aumento lineare di 0,6 nMPb/MCa all’anno fino agli anni ‘80, quando la concentrazione del Pb nell’esoscheletro dei coralli decresce a causa della rimozione del Pb dalle benzine americane. Dall’analisi di dettaglio della distribuzione del Pb attraverso la colonna d’acqua nel Pacifico Settentrionale (Nozaky Y., 1996) risulta che la concentrazione del Pb decresce esponenzialmente dalla superficie al fondo oceanico. Il Pb non presenta nello strato oceanico superficiale ricco di vita il minimo assoluto della concentrazione caratteristica degli elementi che limitano la crescita planctonica, bensì la tipica diminuzione esponenziale della concentrazione con la profondità che è peculiare degli elementi rapidamente rimossi dalla sedimentazione del particellato oceanico (Salomon W., and Förstner U., 1984).
Da alcune analisi riportate da Sahl K., Doe B.R. e Wedepohl K.H. (1978) (tabella T1334-3) si osserva che il Pb è fortemente accumulato nella biomassa marina. Gli organismi che raggiungono le concentrazioni maggiori appartengono al microplancton e al fitoplancton, con 29,5 e 18,3 mg/Kg. Per gli organismi marini si può stimare un fattore di arricchimento rispetto all’acqua marina compreso tra 10+4 e 10+5.
Il contenuto di Pb nelle più comuni rocce sedimentarie (T1334-1), Arenarie, carbonatiche ed Argilliti, è rispettivamente di 12,0, 9,0 e 20 mg/Kg. Il contenuto di Pb, raggiunge le concentrazioni massime nelle Argilliti nere ricche in sostanza organica (30 mg/Kg) e nelle Argilliti di piana abissale (80 mg/Kg), nei quali è principalmente associato agli ossidi di Fe (Yuan-Hui Li, 1982). E’ pertanto ragionevole ritenere che il principale meccanismo di rimozione del Pb dall’acqua oceanica sia la sedimentazione delle argille ed il seppellimento nei sedimenti della sostanza organica.
Sahl K., Doe B.R. e Wedepohl K.H. (1978) riportano ricerche dalle quali risulta che nel corso del processo metamorfico la concentrazione del Pb diminuisce all’aumentare del grado metamorfico,
Figura F1334-1. Distribuzione di
frequenza del Pb (mg/Kg) nei Graniti. (Tratto da Shan K., Doe B.R., e Wedepohl
K.H., 1978).
Figura F1334-2. Distribuzione di
frequenza del Pb (mg/Kg) nelle Rioliti. (Tratto da Shan K., Doe B.R., e
Wedepohl K.H., 1978).
Figura
F1334-3. Diagramma Eh-pH del sistema Pb-O-H-C-S.
(Tratto da Brookis D.G., 1988).
come è per altro evidente dai dati riportati nella tabella T1334-1. La mobilizzazione del Pb nel processo metamorfico sembra comunque fortemente controllata dalla presenza nei fluidi circolanti nelle rocce di Cl, un anione che forma complessi stabili con il Pb e ne aumenta la solubilità. Infatti, come riportano gli autori, le rocce incassanti i giacimenti di Galena risultano fortemente impoverite in Pb e nelle inclusioni fluide associate alle mineralizzazioni si osservano elevate concentrazioni di Cl. La formazione dei giacimenti di Galena è così favorita in quegli ambienti geotettonici dove, come nell’Iglesiente (Cagliari), l’assetto geostrutturale comporta una ingente circolazione di acque marine attraverso le rocce crostali.
In letteratura sono riportate numerose ricerche condotte sul contenuto di Pb nei suoli dei paesi dell’emisfero australe (Davies B.E., 1992. Adriano D.C., 1986, Andrew J.E. et al., 1996. Sposito G., 1989). Queste indicano che la concentrazione di questo elemento è massima negli strati superficiali del suolo e diminuisce con la profondità. La concentrazione è massima negli orizzonti superficiali dei suoli argillosi o ricchi di sostanza organica. A seguito della rimozione del Pb dalle benzine Americane si osserva una progressiva diminuzione della concentrazione del Pb negli orizzonti superficiali dei suoli forestali determinata dalla lisciviazione verso la falda (Andrew J.E. et al., 1996).
La solubilità del Pb nelle soluzioni circolanti dei suoli può essere controllata dalla precipitazione di fasi minerali insolubili, la cui precipitazione dipende dalle condizioni di Eh-pH (figura F1334-3). I principali minerali che possono controllare la solubilità del Pb (Alloway B.J., 1992. Brookins D., G., 1987) sono gli idrossidi Pb(OH)2, l’Anglesite PbSO4, la Cerussite PbCO3, l’Idrocerussite Pb3(CO)3(OH)2, il PbPO4 e la Galena PbS. Nella figura F1334-3 è riportato il diagramma Eh-pH del sistema Pb-O-C-S, che pur trascurando l’importante ruolo della precipitazione del fosfato di Pb nel rimuovere dalla soluzione circolante del suolo, fornisce una descrizione qualitativa soddisfacente del comportamento del Pb in condizioni di terreno, ed in buon accordo con i risultati di indagini condotte in campagna quali riportate da Davies B.E. (1992). La concentrazione del Pb+2 supera le 10^-6 M solo a pH inferiori ad 1. In ambiente riducente la concentrazione del Pb è controllata dalla precipitazione dei solfuri. Nell’ambiente ossidante tipico delle falde e dei suoli ben ossigenati la concentrazione del Pb può essere controllata a pH inferiori a 5 dall’Anglesite o, a pH superiori a 5, dalla Cerussite. Il Pb, come tutti gli elementi di transizione bivalenti forma complessi dotati di elevata stabilità con i gruppi lo ione Cl.
La presenza di Cl nella soluzione circolante del suolo può così diminuire l’attività dello ione Pb+2 innalzandone la solubilità, come osservato per altri elementi di transizione bivalenti: Ni, Cu, e Cd (Doner H.E., 1978. Shas V. M., et al., 1979. ). Il Pb, come gli altri elementi di transizione bi- e trivalenti può essere adsorbito sui minerali delle argille e sulla sostanza organica. L’adsorbimento dei metalli di transizione bivalenti sui suoli e sui suoi componenti chimici è solitamente ben descritto
Tabella T1334-3. Concentrazione del Pb (mg/Kg) negli organismi marini.
(Tratto da Shan K., Doe B.R., e Wedepohl
K.H., 1978).
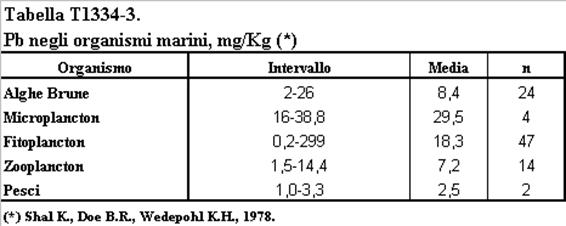
Tabella T1334-4. Parametri
dell’adsorbimento dei metalli di transizione bivalenti sui principali
componenti del suolo. (Elaborato da Bodek I. et al., 1988).
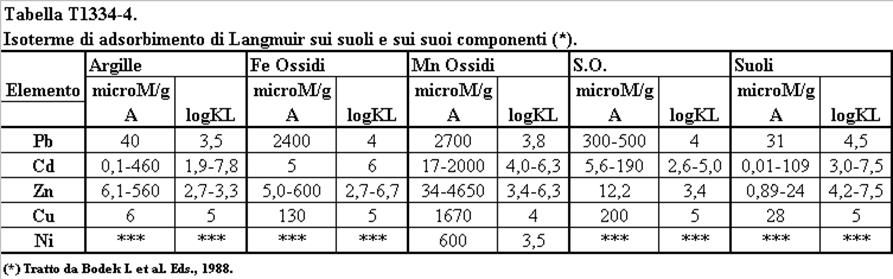
dall’isoterma di Langmuir (Schor J. L., 1996. Stumm W. and Morgan J.J., 1996. Sposito G., 1989). L’isoterma di Langmuir descrive l’adsorbimento nei semplici termini della costante di reazione del metallo con la superficie dell’adsorbente (KL) e della capacità di adsorbimento massima (A) di questa. Come è noto, i parametri della costante di adsorbimento di Langmuir dipendono dalle condizioni sperimentali in cui si effettuano le prove di adsorbimento e non sono estrapolabili con esattezza ad a condizioni sperimentali diverse per pH, forza ionica, composizione della soluzione elettrolitica. Solitamente le isoterme di Langmuir vengono determinate a pH compresi tra 4 e 5 e in presenza di una concentrazione di CaCl2 di 0,01 M/l, condizioni ritenute rappresentativa dei suoli di vaste aree geografiche. Bodek I. et al. (1988) compendiano le isoterme di adsorbimento pubblicate in letteratura per i principali elementi di transizione in traccia (T1334-4). E’ evidente dalla costante di complessazione del Pb con i principali costituenti del suolo che all’adsorbimento del Pb possono partecipare le argille, gli ossidi di ferro di manganese e la sostanza organica. Particolare attenzione è stata dedicata il letteratura all’adsorbimento dei cationi a sulle argille.
In letteratura è riportata una vasta bibliografia sui siti di adsorbimento dei cationi sui minerali argillosi (Bolth G. H., Summer M. E. and Kamphorst A., 1963. Brouwer E., et al., 1983. Cremers A et al., 1988. Ewans D.W. et al., 1983. Francis C. W., and Brincley F.S., 1976. Grutter A., Von Gunten R., and Rossler E., 1986. Hill D.E., and Sawhney B.L., 1969. Klobe W.D., and Gast R.G., 1970. Komarneni S., 1978. Le Roux J., Rich C.I., 1969. Rich C.I., and Blak W.R., 1964. Sawhney B. L., 1965. Sawhney B.L., 1970. Sawhney B. L., 1972. Ziper C., Komarneni S., and Baker E.D., 1988). I minerali argillosi presentano tre principali siti di adsorbimento nei confronti dei metalli:
1) i siti planari delle superfici esterne dei cristalli o siti “p poco selettivi”;
2) i siti planari posti negli interstrati aperti a 14Å di Vermiculiti e Smectiti o siti “p moderatamente selettivi”;
3) i siti ubicati nelle zone a cuneo che fanno da transizione tra i domini cristallografici dove la distanza basale è aperta a 14 Å ed i domini illitici dove la distanza basale è chiusa a 10 Å, o siti “i-e altamente selettivi”.
I siti planari “p” delle superfici esterne dei cristalli ed i siti “p” degli interstrati aperti a 14 Å sono poco selettivi nei confronti dei metalli di transizione bivalenti, ed il metallo ivi adsorbito può essere facilmente scambiato da un cationi idrati come il Ca o il Mg. I siti delle zone che fanno da transizione tra i domini dove il reticolo cristallino delle Illiti è inalterato e chiuso a 10 Å ed i domini cristallografici dove l’Illite è alterata in Vermiculite o Smectite possono formare con i cationi dotati di basso potenziale ionico e bassa energia di idratazione quali Rb, Cs, K, Pb e Cd legami molto forti, le cui energie aumentano con la carica superficiale del minerale argilloso e sono nell’ordine di grandezza dei legami ionici (Brouwer E., et al. 1983). I siti altamente selettivi rappresentano di norma una piccola frazione della capacità di scambio cationico delle argille, variabile con il loro grado di alterazione in minerali secondari, (Bolth G. H., Summer M. E. and Kamphorst A., 1963. Cremers A et al., 1988) e sono efficaci nel controllare la solubilità dell’elemento solo quando questo è presente nel suolo in tracce (Bolth G.H. Ed., 1979).
Davies B.E., (1992) Compendia una ricca raccolta dei risultati delle indagini dirette condotte sul comportamento del Pb in suoli contaminati. I risultati ottenuti indicano un comportamento del Pb fortemente differente in funzione del pedoambiente. Alcuni studi condotti sulle forme chimiche del Pb presente nella soluzione circolante del suolo provano che nei suoli carbonatici la sua concentrazione è controllata dalla solubilità della Cerussite PbCO3 e dall’Idrocerussite Pb3(CO)3(OH)2. In suoli non carbonatici la solubilità del Pb è talvolta controllata dal Anglesite PbSO4, dall’adsorbimento sugli ossidi di manganese o sulla sostanza organica. Gli studi condotti mediante frazionamento chimico delle forme in cui il Pb è presente nei suoli riportati da Davies B.E. (op. cit.) indicano che in alcuni suoli la sostanza organica è il principale componente del suolo che fissa il Pb in forme non scambiabili, chelandolo attraverso i gruppi carbossilici degli acidi umici. In altri suoli la sostanza organica non ha un ruolo statisticamente significativo nel limitare la quota del Pb scambiabile, che è determinata principalmente dalla la capacità di scambio cationico. La composizione mineralogica del suolo, in particolare quella dei Fillosilicati sembra un fattore importante nel determinare l’influenza della tessitura sul comportamento del Pb e degli altri elementi di transizione con basso potenziale ionico. Nel suolo della Scozia, fortemente lisciviato, la solubilità del Pb risulta infatti inversamente correlata alla frazione argillosa fine, presumibilmente composte da Vermiculiti o Smectiti, minerali argillosi poveri di siti altamente selettivi nei confronti del Pb. La frazione tessiturale che fissa il Pb è quella limosa, presumibilmente composta da Illiti e Idromiche, ricche di siti di adsorbimento altamente selettivi. Dai risultati dei frazionamenti riportati da Davies B.E., (1992) risulta che gli ossidi di manganese risultano per lo più di scarso rilievo nel fissare il Pb.
Il risultato è in disaccordo con gli studi sull’adsorbimento del Pb sugli ossidi di manganese e di ferro, dai quali risulta che l’energia con cui il Pb è legato a questi composti minerali non è molto diversa da quella con cui è legato alla sostanza organica (Bodek I. et al., 1988). L’osservazione è inoltre in disaccordo con la forte correlazione osservata tra contenuto di Pb, Cd, Zn, Cu, Ni e Co dei sedimenti e il contenuto in ossidi di Fe e Mn dei sedimenti fluviali riportata da molti autori in (Nowland G. A., 1976. Carpenter et al., 1978. Gibbs R.J., 1977. Teraoka H. and Kobayashi J., 1980). La forte correlazione tra contenuto di elementi di transizione in traccia bivalenti e contenuti in ossidi di Fe e Mn induce a ritenere che i sesquiossidi siano le principali fasi che nel corso del processo pedogenetico occludono gli elementi liberati dall’alterazione dei reticoli cristallini dei minerali primari. E’ stato comunque osservato (Salomons W., Förstner U., 1984) che nei sedimenti fluviali la ripartizione degli elementi di transizione in traccia bivalenti nelle diverse forme è varia in funzione del bacino idrografico (F1334-4). E’ evidente dalla figura F1334-4 che i sedimenti fluviali provenienti da bacini idrografici fortemente industrializzati, riportati sulla sinistra della figura, si caratterizzano per trasportare elevate quantità di Mn e Fe ed una elevata percentuale di elemento scambiabile, legato alla sostanza organica ed occluso nei sesquiossidi. I sedimenti fluviali provenienti da bacini scarsamente industrializzati, posti alla sinistra della figura, sono al contrario caratterizzati dal minori quantità di Mn e Fe, una percentuale trascurabile di elemento scambiabile e una percentuale preminente di elemento occluso nei minerali primari. E’ ragionevole ritenere che le diverse forme assunte dal e dagli
altri elementi di transizione in traccia bivalenti nei sedimenti fluviali riflettano le forme che gli elementi hanno nei suoli dei rispettivi bacini idrografici sia le differenze dei processi erosivi in atto nei diversi bacini.
Davies B.E., (1992) e Adriano D.C. (1986) compendiano una ricca raccolta di ricerche condotte sulla biodisponibilità del Pb presente nel suolo. Nell’intervallo di concentrazione del Pb comunemente osservato nei suoli si osserva una correlazione lineare tra la concentrazione dell’elemento nel suolo e quella nel vegetale. Secondo la rassegna critica sulla biodisponibililità degli inquinanti di Baes C.F et al. (1984) la concentrazione del Pb negli organi vegetativi e quiescenti delle piante è mediamente rispettivamente il 4,5% ed il 0,9% della concentrazione dell’elemento nel suolo. Considerando un contenuto medio del Pb nei suoli americani di 19 mg/Kg il contenuto medio di Pb nei foraggi può essere grossolanamente stimato di 0,85 mg/Kg. Il Pb raggiunge la sua massima concentrazione nelle radici dei vegetali ed è traslocato nella porzione apogea con difficoltà (Davies B.E., 1992). Quando nel suolo raggiunge elevate concentrazioni, il Pb è immobilizzato dalla suberificazione dell’epidermide radicale. La capacità di assimilare il Pb e traslocarlo negli organi apogei varia da specie a specie e risente della fase vegetativa. Tra le proprietà del suolo che limitano l’assimilazione del Pb il pH non sembra avere grande rilievo, in quanto risulta essere assimilato anche nei suoli carbonatici. Il principale fattore che sembra controllare la biodisponibilità del Pb è la presenza di cationi che competono nell’adsorbimento del metallo sulle argille.
Come riporta Davies B.E., (1992) la deposizione di Pb al suolo è per il Nord America compresa tra 7.1 e 2050 mg·m2·a-1 , con una media di 424 mg·m2·a-1 mentre per l’Europa compresa tra 8.7 e 53.6 mg·m2·a-1 con una media di 19 mg·m2·a-1. Nel computo della assimilazione del Pb nei vegetali non si può quindi, come osservano Sheppard S.C. et al. (1992), trascurare l’assorbimento fogliare. In un rilievo condotto sulla concentrazione del Pb nei vegetali della Norvegia Meridionale (Bertelsen B.O., et al. 1995), si osserva che la concentrazione del Pb in un’ampia gamma di specie e organi vegetali epigei (n=18) diminuisce dal 1982 al 1992 dl un valore medio di 9,3 +/- 172% al valore di
Figura F1334-4. Forme del Fe, Mn, Zn, Cu,
Ni, Pb e Cd nei sedimenti trasportati dai fiumi. (NH4Ac=solubile+scambiabile;
NH2OH*HCl=occluso negli ossidi di manganese; H2O2/HCl=estraibile
con attacco acido forte; resistant=estraibile in acido fluoridrico). (Tratto da
Salomon W. e Förstner U., 1984).
4,6 +/-122% mg/Kg. Negli stessi anni la deposizione del Pb diminuisce da 12,2+/-36% a 3.9+/-29% mg·m2·a-1 (n=4). Nonostante la forte diminuzione di deposizione di Zn, Cd e Cu, per questi elementi non si osserva, come per il Pb, una significativa diminuzione della concentrazione nei tessuti vegetali epigei.
Studi isotopici condotti sull’origine del Pb nel sangue umano (Lee Robert C., et al., 1995. Lewandosky T.A., and Forlsund B. L., 1994) indicano che questo può avere origini molto diverse. In aree poco contaminate la principale sorgente del Pb presente nel sangue è il consumo di derrate alimentari e l’ingestione di suolo. In aree fortemente contaminate, come in prossimità degli stabilimenti siderurgici, o gli ambienti urbani, la principale fonte attraverso cui il Pb è assimilato è l’ingestione involontario del particellato argilloso su cui questo elemento è fortemente arricchito (Sheppard S. C., 1995. Sheppard S.C., et al., 1992). Gli stessi studi, nell’alea del limitato numero di campioni considerati, indicano che circa l’1% della popolazione infantile delle comunità urbane di città occidentali raggiunge livelli di Pb ematico ai quali possono manifestarsi effetti neurotossici.
Adriano D.C. (1986) compendia una raccolta di studi sulla fitotossicità del Pb. Prove in coltura idroponica indicano che la sensibilità al Pb+2 varia con la specie. Il limite massimo di Pb tollerabile per lo sviluppo del vegetale è di 50 mg/l per il pomodoro, 25 mg/l per la bietola, 30mg/l per il pisello, corrispondenti per la bietola ad una concentrazione di 35 mg/Kg. Le concentrazioni di Pb nel suolo cui si manifesta tossicità sui vegetali sono molto variabili in funzione del pedoambiente e dell’essenza: per il riso è stata segnalata fitotossicità per concentrazioni di Pb di 50-2000 mg/Kg su alcuni suoli e 400-500 mg/Kg su altri. Il Pb blocca la respirazione mitocondriale, altera il trasporto di elettroni nella fotosintesi, in altri termini, inibisce il metabolismo vegetale. Per il grano e la soia la fotosintesi diminuisce linearmente con la concentrazione di Pb nei tessuti. Quando il contenuto di Pb raggiunge nei tessuti del grano i 60 mg/Kg la perdita fotosintetica è del 20%, mentre per la soia è del 10%. Per la biomassa vegetale della Norvegia Settentrionale si può cautelativamente stimare quindi una diminuzione della fotosintesi compresa tra il 4 ed il 1,4%.
Sull’organismo umano non si osservano effetti tossici per concentrazioni ematiche inferiori a 20g/100 ml (Lilia A.A., and Badillo F., 1991). Tra i 20 ed i 40 g/100ml si osserva anemia, mentre sopra tale soglia si osservano effetti neurotossici ed una correlazione negativa tra la concentrazione di sangue ematico e quoziente intellettivo. Elevati valori di sangue ematico si osservano in particolare nella popolazione infantile delle comunità siderurgiche, in prossimità di stabilimenti per lo smaltimento delle batterie esauste e in ambiente metropolitano. Lilia A.A., and Badillo F., (1991) segnalano che da un recente studio epidemiologico condotto nei quartieri popolari di Città del Messico il 30% dei bambini ha problemi di apprendimento. Solo il 17% del campione ha livelli ematici inferiori a 20g/100ml, il 23% ha livelli ematici compresi tra 21 e 39 g/100ml, mentre il 30% ha livelli ematici sopra i 60 g/100ml.
La principale fonte mineraria da cui è estratto il Pb è la Galena. La produzione annua mondiale degli ultimi 30 anni è stata di circa 32 milioni di tonnellate (Adriano D.C., 1986). Il 60% del Pb è impiegato nella industria automobilistica, come componente delle leghe e come additivo antidetonante delle benzine. Il rimanente 40% è impiegato nella produzione delle batterie. La maggiori sorgenti di inquinamento del suolo sono quindi le emissioni in atmosfera determinate dalle attività siderurgiche e dal traffico veicolare, stimate complessivamente in ragione di 400000 tonnellate/anno (Adriano D.C., 1986).
Il
Cd ha un potenziale ionico simile a quello del Pb e di conseguenza un
comportamento simile nel ciclo petrogenico. La concentrazione del Cd cresce con
il contenuto di silice delle rocce ignee, è minima per le rocce ultrafemiche
(0,05 mg/Kg), massimo nelle rocce basiche (0,2 mg/Kg) e massimo nelle rocce
acide (0,15 mg/Kg). In occorrenza delle dorsali medio-oceaniche in
corrispondenza delle quali si ha la fusione parziale delle peridotiti del
mantello, il Cd, dotato di basso potenziale ionico, si concentra nel fuso
parziale della peridotite che da luogo al magma basaltico. La cristallizzazione
dei minerali ricchi in Fe e Mg del magma basaltico, per lo più poveri di
elementi a basso potenziale ionico, non
è efficiente nel rimuovere il Cd dal fuso, che raggiunge così le concentrazioni
massime nelle rocce acide. Il Cd ha un potenziale ionico simile a quello del K,
ed è pertanto seguendo i principi generali della cristallochimica (Faure G.,
1992) è corretto ritenere esso vicarii questo elemento maggiore nei
K-Feldspati, nei Fillosilicati e nei Plagioclasi, occupando preferenzialmente i
siti cristallografici a coordinazione 8 o 12. Il Cd è un elemento calcofilo e
può raggiungere concentrazioni molto elevate nei solfuri, in particolare nella
Galena, e nella Sfalerite, dove può facilmente vicariare il Pb e lo Zn. I
solfuri delle rocce, ed in particolare la Pirite, non sono comunque presenti in
quantità sufficienti ad occludere tutto l’elemento presente nel magma, che è
pertanto in buona parte ospitato nei reticoli cristallini dei minerali primari
di K e delle Apatiti (Whedwpohl K.H., 1969).
Le
forme che il Cd ha nei sedimenti trasportati dai corsi d’acqua provenienti da
bacini idrografici che ospitano una fiorente industria, sono riportate sulla
sinistra della figura F1334-4. Le
forme prevalenti del Cd sono quelle solubili, scambiabili ed occluse negli
ossidi di manganese. Nella colonna d’acqua marina la concentrazione del Cd ha
il valore minimo nei primi 200m, indicando che nello strato oceanico ricco di
vita è fortemente accumulato dagli organismi planctonici. Nello strato
sottostante ha una concentrazione maggiore e costante, indicando che non è
efficientemente rimosso
Figura F1335-1. Diagramma Eh-pH del
sistema Cd-S-C-O-H.
Tabella T1335-1. Costante di
selettività delle Ca-Argille verso Pb, Cd, Zn e Cu.
(Elaborato da Bolth G.H., 1976).
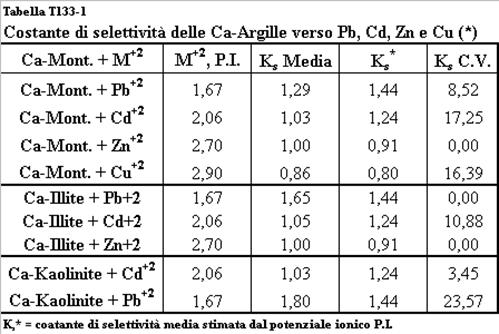
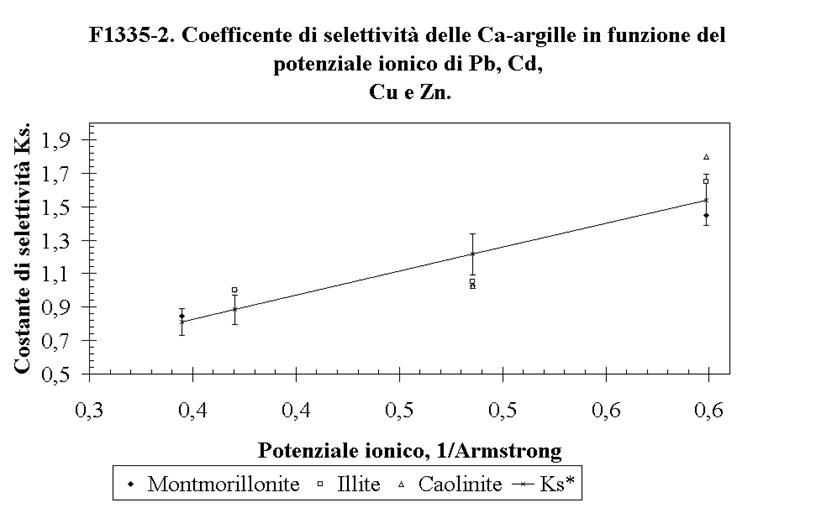
Figura F1335-2. Rapporto tra
potenziale ionico dell’elemento di transizione bivalente ed la costante di
selettività delle Ca-Argille. (Elaborato da Bolth G.H., 1976).

Figura F1335-3. Stabilità dei
complessi dell'acido acetico con alcuni metalli pesanti bivalenti.
Figura F1335-4. Rapporto tra pH del
suolo e costante di Langmuir KL.
(Tratto da Shulte A. e Beese F., 1994).
dall’acqua marina a causa della
sedimentazione della sostanza organica e delle argille. Il tempo di residenza
del Cd nell’oceano è quindi presumibilmente lungo rispetto a quello del Pb,
rapidamente rimosso dalla sedimentazione della sostanza organica e delle
argille. La concentrazione del Cd nelle rocce sedimentarie è bassa nelle
Argilliti di piana abissale (0,5 mg/Kg), ed è maggiore nelle Argilliti di
piattaforma continentale (1 mg/Kg). Bassa è la concentrazione del Cd nelle
rocce carbonatiche, 0,05 mg/Kg. La distribuzione del Cd nelle rocce
sedimentarie suggerisce che il principale meccanismo di rimozione del Cd dalle
acque oceaniche sia la sedimentazione delle argille. La concentrazione del Cd è
molto elevata nel carbone, 1,3 mg/Kg, indicando che questo elemento è fortemente
accumulato dalla vegetazione terrestre. La concentrazione del Cd nella crosta
terrestre è di 0,11 mg/Kg.
La
concentrazione del Cd nei suoli Russi (Vinogradov, 1959) risulta da rilievi
condotti nella prima metà del ‘900 di 0,1 mg/Kg, indicando un fattore di
arricchimento del suolo rispetto alla crosta terrestre uguale a 1,0. Da rilievi
condotti più recentemente (Adriano D.C., 1990) la concentrazione del Cd risulta
di 0,26 mg/Kg nel Nord America, 0,7 mg/Kg nel Regno Unito e in media di 0,62
mg/Kg nel mondo. L’aumento della concentrazione del Cd nei suoli è
principalmente dovuta alle attività antropiche degli ultimi 50 anni (Adriano
D.C., 1990. Alloway B.J., 1990). Nel
suolo la solubilità del Cd può essere controllata dalla precipitazione di fasi
insolubili che dipendono dalle condizioni di Eh-pH, (figura F1335-1). La solubilità del Cd è
limitata in condizioni riducenti dalla precipitazione dei solfuri. In ambiente
ossidante la solubilità del Cd è controllata dal pH. A pH superiori ad 8,
tipici dei suoli carbonatici ed alcalini si ha la precipitazione del carbonato,
degli ossidi e degli idrossidi. Nell’ambiente ossidante tipico delle acque
superficiali e di falda il Cd è fortemente solubile a pH inferiori ad 8. Come
per tutti gli altri elementi di transizione bivalenti, il Cd forma complessi
stabili con il Cl che ne possono innalzare la solubilità anche a pH alcalino o
in ambienti riducenti (Doner H.E., 1978).
La
solubilità del Cd, come quella di
Rb, Cs, K, e Pb, avendo un modesto potenziale ionico
e bassa energia di idratazione, può essere adsorbito nei siti altamente selettivi tipici delle
Illiti e delle Idromiche (Ziper C., Komarneni S., e Baker D. E., 1988, Sawney)
nei quali è legato con energie prossime a quelle dei legami ionici. I siti
planari dei minerali argillosi, non sono selettivi nei confronti del Cd, che
può essere facilmente scambiato dal Ca. Per queste argille il coefficiente di
selettività nell’adsorbimento di questi due cationi è infatti mediamente di 1,0
(tabella T1335-1 e figura F1335-2). La solubilità del Cd può
essere inoltre controllata dalla complessazione con i gruppi carbossilici dei
composti organici, di cui, a titolo di esempio,
sono riportate le costanti di stabilità dell’acido acetico nella tabella
T1335-2, F1335-3. All’adsorbimento
del Cd possono ovviamente partecipare anche gli ossidi di Fe e Mn (tabella T1334-4). Sculte A. e Beese F. (1994)
studiano 16 campioni di suolo Israeliani e Tedeschi e trovano che
l’adsorbimento del Cd sul suolo può essere descritto dall’isoterma di Langmuir.
La costante di complessazione KL dell’isoterma di Langmuir dipende linearmente
dall’attività dello ione idronio e dal componente minerale che ne tampona
l’attività (figura F1335-4), mentre
il massimo di adsorbimento A aumenta con il contenuto di argilla. Holm P.E. et
al. (1996) osservano che la soluzione circolante dei suoli agrari carbonatici,
tamponati a pH 8 dalla Calcite, la concentrazione del Cd può raggiungere valori
superiori a 10 volte quella prevedibile dalla precipitazione del l’Octavite,
CdCO3,
probabilmente a causa della presenza nella soluzione circolante di sostanza
organica solubile. McBride M.B. et al.
(1981) osservano che l’adsorbimento del Cd su un campione di suoli
rappresentativo dell’America
Nord-Occidentale, dove il pH è varia tra 4,8 e 7,3 mentre il contenuto di
argilla varia tra il 2 e il 52%, è ben
descritto dall’isoterma di Langmuir. Impiegando una soluzione di riferimento
10^-5 M osservano che la quantità di Cd legato alla
frazione solida è inversamente proporzionale al Ca scambiabile. La frazione di
Cd adsorbita sul suolo è a sua
volta inversamente proporzionale
alla concentrazione del Cd nel grano cresciuto sul suolo contaminato dal Cd.
Gli autori concludono pertanto che il principale fattore di controllo della biodisponibilità
del Cd antropico è la reazione di scambio tra Cd e Ca sulle argille. Come
riporta Alloway (1997) molti autori trovano una correlazione lineare
significativa tra contenuto di Cd nel vegetale e nel suolo. Secondo la rassegna
critica di Baes C.F. et al. (1984) sui fattori di trasferimento suolo-pianta,
la concentrazione del Cd antropico nelle porzioni vegetative e quiescenti dei
vegetali è mediamente il 55% ed il 16% della concentrazione dell’elemento nel
suolo, indicando un forte accumulo dell’elemento nella biomassa vegetale. Per i
suoli del Nord America dove il contenuto di Cd è mediamente di 0.35 mg/Kg si
può stimare il contenuto medio di Cd nei foraggi di 0,19 mg/Kg e nella granella
dei cereali di 0,21 mg/Kg. Per gli elementi di cui si registrano elevati tassi
di deposizione dall’atmosfera, come osservato da Sheppard S.C. et al (1992) per
valutare la concentrazione nella
biomassa vegetale oltre all’assorbimento radicale andrebbe considerato
l’assorbimento fogliare. Bertelsen B.O. et al. (1995) la concentrazione del Cd
in un ampio campione di tessuti vegetali della Norvegia Meridionale, soggetta
ad una deposizione atmosferica di 2,9+/-24% mg·m-2·a-1. La
concentrazione del Cd risulta di 0,29+/-94% mg/Kg. La concentrazione media del
Cd nella biomassa vegetale non cambia significativamente dieci anni dopo, nel
1992, quando la deposizione al suolo di Cd è scesa a 0,09+/-5% di mg·m-2·a-1, suggerendo che il Cd manifesti una bassa
assimilabilità fogliare.
Le
concentrazioni di Cd nei tessuti vegetali che determinano un decremento del 25%
della fotosintesi variano sensibilmente secondo la specie (Adriano D.C., 1986).
Su un campione di 19 essenze esse sono risultate comprese tra 160 mg/Kg e 3
mg/Kg, con una media di 56+/86% (n=19) mg/Kg. La dose giornaliera assimilabile
dalla popolazione di Cd consigliata dalla FAO è di 0,060.07 mg/g. La dose assimilata dalla
popolazione mondiale, è compresa tra
0.025 e 0.075 mg/g (Alloway B.J., 1997). L’assimilabilità del Cd ingerito con l’alimentazione
è nei mammiferi modesta; solo 1-2% per il ratto, 0,5-3% per la scimmia, 16% per
le vacche, 3-8% per gli uomini (Ragan H.A. e Mast T.J., 1990). I minerali
argillosi non diminuiscono la biodisponibilità del Cd antropico ingerito
accidentalmente con il suolo (Sheppard S.C. et al., 1994). L’assimilabilità del Cd inalato è doppia rispetto a quella del Cd ingerito
(Ragan H.A. e Mast T.J., 1990). Tra gli effetti sanitari dell’intossicazione da
Cd vi è la diminuzione della fertilità maschile (op. cit.), un fenomeno che,
come noto, affligge le popolazioni urbane, gli allevamenti bovini e che è, con
la caccia, tra le cause della diminuzione del numero di branchi di Balene.
Le
principali sorgenti d’emissione antropica di Cd nell’ambiente sono: i fertilizzanti
fosfatici, i fanghi di depurazione e le attività inerenti l’estrazione e la
lavorazione di minerali. Il Cd, come Pb, Bi e Sb è contenuto già in origine nella fosforite,
la roccia sedimentaria organogena dalla quale vengono prodotti i fertilizzanti
fosfatici (Wedepohl, 1969). Infatti è riscontrata una correlazione diretta tra
l’accumulo di P e Cd sulla superficie dei suoli a testimoniare la pericolosità
di un indiscriminato
utilizzo di tali fertilizzanti
(Alloway B.J., 1997). Il suo contenuto nei fanghi di depurazione utilizzati in
agricoltura dipende dalla tipologia del rifiuto e dall’estensione del centro
urbano che li produce. Le acque di scarico impiegate nelle attività estrattive
di alcuni minerali quali, in particolare, la Sfalerite (ZnS) e la Smithsonite
(ZnCO3) hanno
tenori in Cd decisamente inquinanti (540 mg/kg). Un’ulteriore conseguenza
causata dai processi di fusione dei minerali sopracitati è rappresentata
dall’immissione nell’atmosfera del Cd, stimata di 5510^8 g/a (Adriano
D.C., 1986) e, quindi, di una successiva deposizione la cui concentrazione
dipende dalla distanza dalla sorgente d’emissione.
Lo
Zn+2 ha un potenziale ionico di 2,7 Å-1, simile a quello del Fe+2 (2,62 Å-1) di cui è vicariante nelle strutture cristalline dei
minerali primari. La sua concentrazione . nelle rocce ignee, è minima nelle peridotiti (15 mg/Kg), massima nei
basalti (90 mg/Kg) e minima nelle rocce acide (15 mg/Kg). Esso si
concentra nel fuso basaltico prodotto
dalla fusione parziale delle rocce peridotitiche del mantello. Dal magma
basaltico, a differenza di Pb e Cd, ioni troppo grossi per vicariare il Fe+2, è efficacemente rimosso dalla
cristallizzazione frazionata dei minerali di Femici, principalmente dai
Pirosseni ed dagli Anfiboli. Esso inoltre può vicariare il Fe+2 nella Pirite, dove raggiunge elevate
concentrazioni. Nel magma acido rimangono perciò modeste quantità di Zn, che
vengono ad essere catturate come elemento in traccia prevalentemente nella
Biotite e negli altri minerali femici che, come i pirosseni, talvolta si
osservano nelle rocce acide (Wedepohl, 1969). La sua concentrazione nel suolo
può essere controllata dalla precipitazioni di solfuri in ambiente riducente.
In ambiente ossidante è altamente solubile a pH inferiore ad 8, mentre a pH
inferiori precipita come ossido (Brookis D.G., 1988). Come gli altri elementi
di transizione bivalenti la sua concentrazione nella soluzione circolante nel
suolo può essere controllata dall’adsorbimento sui colloidi organici e le
argille (T1334-4). Da queste ultime,
avendo un potenziale ionico piuttosto elevato e una energia di idratazione
relativamente alta, non fissato nei siti specifici dei bordi di alterazione
delle Illiti e delle Idromiche. Come per
gli altri elementi di transizione bivalenti molti autori riportano una forte
correlazione tra il contenuto di Zn e quello di Fe e Mn nei sedimenti fluviali.
Le forme che assume in questi dipendono dal bacino idrografico (Figura F1334-3). Nei bacini delle aree
industrializzate prevalgono le forme solubili, scambiabili e legate agli ossidi
di Mn mentre nei bacini idrografici delle aree poco industrializzate prevalgono
le forme occluse nei minerali primari. Nell’oceano, lo Zn, come il Cd, si
comporta da fattore limitante la crescita planctonica nei primi 200 m di
spessore oceanico e nello strato sottostante raggiunge una concentrazione
elevata e costante, indicando che è rimosso dalla sedimentazione della sostanza
organica con scarsa efficacia. Il suo tempo di residenza nell’oceano è di 10^310^4 a (Whitfield
M., 1981). La concentrazione dello Zn nelle rocce sedimentarie è massima nelle
Argilliti di piana abissale (165 mg/Kg), dove è associato prevalentemente agli
ossidi di manganese, intermedia nelle Argilliti ricche in sostanza organica
(100 mg/Kg) e bassa nelle Argilliti povere di carbonio organico (10 mg/Kg). Nei
carbonati ha basse concentrazioni, 1,7 mg/Kg. Dalla distribuzione dello Zn
nelle rocce sedimentarie è ragionevole ritenere che i principali meccanismi di
rimozione dalle acque oceaniche sia il seppellimento della sostanza organica e
la sedimentazione delle argille. II contenuto di Zn nel carbone è di 1,7 mg/Kg
indicando che questo elemento rispetto al Pb ed al Cd è meno fortemente
bioaccumulato dalla vegetazione continentale rispetto al Pb, del Cd e dell’As.
Tra
le sorgenti di inquinamento del suolo vi è lo Zn è contenuto nei fertilizzanti
sia organici che inorganici, come il solfato di zinco (ZnSO4), l’ossido di zinco (ZnO) ed il
complesso Zn-NH3 utilizzati in agricoltura. Altre fonti
inquinanti possono essere rappresentate dalle polveri dei combustibili fossili,
dalla fusione dei metalli non ferrosi e dai fanghi di depurazione.
Particolarmente variabile è il contenuto di Zn nei fanghi di depurazione che
risulta spesso superiore ai livelli di fondo dei suoli.
Il
Cu ha un potenziale ionico più elevato simile a quello dello Zn e una analogo
comportamento nel ciclo petrogenico. La sua concentrazione nelle rocce ignee è
massima nelle rocce basiche ed intermedia nelle rocce basiche e acide. E’ stata
osservata una significativa correlazione tra il contenuto di Cu e di S nelle
rocce basiche, indicando che la cristallizzazione della Pirite nel magma
basaltico rimuove quantità significative di Cu dal fuso silicatico. Tra Pirite
e Calcopirite vi è infatti una buona miscibilità allo stato solido (Wedepohl K.
H., 1969).
Nei
suoli la sua solubilità è controllata in ambienti riducenti dalla
sedimentazione dei solfuri, in ambienti ossidanti il Cu è solubile a pH
inferiori a 6, mentre a pH superiori la sua solubilità è controllata dalla
precipitazione di ossidi, idrossidi, carbonati e solfati (Brookis D.G, 1988).
Avendo elevato un elevato potenziale ionico e una elevata energia di
idratazione non è fissato in siti altamente selettivi delle Illiti. Esso forma
legami particolarmente stabili con gli ossidi di Mn e i composti umici (T1334-4). Secondo Baes C.F et al.
(1984), la concentrazione del Cu antropico nei tessuti vegetativi e quiescenti
è rispettivamente il 40 ed il 25% della concentrazione dell’elemento nel suolo.
Le forme del Cu rilevate nei sedimenti fluviali sono, come per gli altri
elementi di transizione diverse a seconda del grado di industrializzazione del
bacino idrografico di provenienza (F1334-3).
Nelle acque oceaniche la concentrazione del Cu è minima in superficie e cresce
linearmente con la profondità. La concentrazione del Cu nelle rocce
sedimentarie indica che il principale meccanismo di rimozione del Cu dalle
acque marine è stata, nel corso delle ere terrestri, la sedimentazione delle
argille e della sostanza organica. Il suo tempo di residenza nell’acqua
oceanica è stimato di 10^3 anni (Whitfield M., 1981).
La
fonte più rilevante di Cu nei suoli agricoli è rappresentata da alcuni suoi
composti, utilizzati come fertilizzanti, quali: i chelati, capaci di mantenere
il Cu biodisponibile per i vegetali, il solfato di rame idrato (CuSO4 5H2O) contenente il 25,5 % di rame ed alcuni ossidi (CuO,
Cu2O). Di
particolare interesse il loro utilizzo come fungicidi e battericidi nella
protezione sanitaria della vite, poiché efficaci nel combattere la Peronospora,
parassita diffuso in tutti i paesi viticoli del mondo. I preparati “storici”
più efficaci sono le poltiglie cupriche, tra cui la poltiglia borgognona e
quella bordolese sono, da sempre, le più impiegate. La poltiglia borgognona
consiste in una soluzione di solfato di rame idrato neutralizzata con carbonato
di sodio (Na2CO3), mentre quella bordolese è
costituita sempre da solfato di rame idrato, neutralizzato, in questo caso, con
calce spenta Ca(OH)2.Questi composti cuprici amorfi
sotto l’azione dell’acqua piovana carica di anidride carbonica o sotto l’azione
della rugiada liberano ioni rame solubili. Tra i preparati moderni più adottati
vi sono i fungicidi cuprici puri, le poltiglie bordolesi essiccate e
micronizzate, che è sufficiente mettere in sospensione nell’acqua per poterle
utilizzare direttamente nel trattamento della vite, e le miscele
organo-cupriche, ottenute aggiungendo ad uno o più fungicidi organici di
sintesi composti quali l’ossicloruro tetracuprico, il solfato di rame e
l’idrossido di rame. Alti quantitativi di Cu si possono trovare anche nei
fanghi di depurazione e nelle deiezioni degli animali d’allevamento nutriti con
mangimi addizionati in solfato di rame, che viene sempre più impiegato
nell’alimentazione animale, poiché è risultato essere un valido integratore
alimentare.
Gli
apporti più rilevanti di Ni antropogenico provengono dalle emissioni delle
fonderie e dai fanghi di depurazione dove supera lo 0,5 % e rappresenta, con lo
Zn ed il Cu, il metallo pesante che potenzialmente può risultare più
fitotossico.
Particolarmente
ricchi in Co sono i liquami animali e le acque di scarico prodotte dalle
fonderie, che vengono talvolta riversate su vaste aree con conseguente rischio
di contaminazione. Minori quantitativi sono presenti nelle ceneri prodotte
durante la combustione del carbone (specie quello bituminoso) (vedi Tabella
1-6). Un’altra potenziale sorgente inquinante è costituita da un uso eccessivo
dei sali di Co o dei fertilizzanti fosfatici trattati con il Co.
Le
fonti d’inquinamento principali del Cr sono rappresentate dai fertilizzanti
fosfatici, che possono contenere oltre 1000 mgr/Kg di Cr, e dalla deposizione
atmosferica provocata dalle emissioni delle industrie metallurgiche,
specialmente acciaierie o stabilimenti che estraggono e trasformano il Fe.
Minori quantitativi vengono prodotti dagli impianti di riscaldamento, dalle
centrali elettriche e dai cementifici (vedi Tabella T132-1).
2.
Obiettivi della ricerca sono di indagare la mobilità degli inquinanti emessi in prossimità di stabilimenti siderurgici ed identificare i fattori chimici e fisici che la controllano. In particolare si indagano:
1) La distribuzione areale dell’inquinante attorno allo stabilimento;
2) l’innalzamento antropico della concentrazione litogenica dei microelementi inquinanti;
3) la velocità di lisciviazione dell'inquinante;
4) l’importanza della geomorfologia e dell’uso del suolo nel determinare la mobilità degli inquinanti;
5) l’importanza della geomorfologia e dell’uso del suolo nel determinare la biodisponibilità degli inquinanti.
3.
3.1.1. La contea di Cliland, Glasgow, Scozia.
La contea di Cliland è ubicata all’interno di una grande unità tettonica, nota con il termine di “The Midland Valley” o “The Great Valley”, la grande Cameron B.I. e Stephenson D., 1985). Dal Devoniano al Siluriano nella Great Valley si registra la deposizione di Arenarie continentali rosse a festoni tipiche di clima desertico. Ad incominciare dal siluriano la Great Valley viene a trovarsi delimitata da due faglie distensive orientate in direzione E-W e diviene soggetta ad un lento sprofondamento tettonico al quale si accompagna un cambiamento della tipologia dei sedimenti. Dai rilievi posti a nord e a sud della Great Valley delimitati dagli specchi delle faglie distensive è eroso materiale terrigeno, intercalato da sedimenti marini di ambiente poco profondo testimoni di periodiche ingressioni marine. In particolare, nel corso del Carbonifero, quando il Regno Unito era venuto a trovarsi a latitudini equatoriali, le sequenze sedimentarie che colmano la Great Valley sono composte da alternanze ciclotemiche composte da strati di rocce carbonatiche, carbone e limi tipiche del periodico susseguirsi di ambienti deposizionali di mare poco profondo, lacustri e di pianura alluvionale. Nel Triassico all’apertura dell’Oceano Atlantico si associa una fase compressiva con direttrice N-S che determina il sollevamento delle “Higlands”, i rilievi collinari della Scozia Settentrionale. Soggette ad un rapido sollevamento, stimabile nell’ordine di grandezza dei cm/a, l’erosione porta ad affiorare le rocce della crosta continentale superiore, principalmente Scisti Gneiss e rocce granitoidi. Le faglie distensive che delimitano a nord ed a sud il bacino sedimentario della Great Valley sono riattivate in chiave compressiva ed i sedimenti che riempiono la depressione tettonica della Great Valley rimangono ripiegati a formare una anticlinale ad asse E-W pizzicata tra i blocchi continentali convergenti delle Higlands e della Scozia Meridionale. Alla fase compressiva del Cenozoico si associa ovviamente un sollevamento eustatico che porta all’erosione dei sedimenti Cenozoici ed all’affioramento del substrato Paleozoico del riempimento sedimentario della Great Valley.
Nel corso del quaternario in occasione dei periodi glaciali il Regno Unito Settentrionale e Centrale è stato periodicamente coperto da ghiacciai continentali dello spessore di 1000-2000 m. L’ultima glaciazione del Pleistocene, denominata “glaciazione flandriana”, rimodella completamente la superficie topografica del Regno Unito fino alle regioni centrali e traccia un sistema di valli glaciali ampie diverse decine di Km e solcate da Drumlins, rilievi collinari lunghi qualche chilometro ed alti qualche centinaio di metri, orientati parallelamente alla linea di deflusso delle lingue del ghiacciaio Flandriano. I Ghiacciai flandriani erodono quasi completamente i sedimenti quaternari deposti nel precedente periodo caldo, ed al loro ritiro ricoprono la Scozia Centrale e Settentrionale di depositi glaciali, periglaciali e fluvioglaciali. Sulla superficie di deglaciazione si imposta l’attuale reticolo idrografico della Great Valley, che segue le ampie valli glaciali insinuandosi tra i Drumlins. A causa dello scioglimento dello spesso ghiacciaio flandriano, nell’Olocene le faglie che delimitano a Nord (Higland Boundary Fault) e a Sud (Sutern Upland Fault) sono state riattivate e la Great Valley è divenuta soggetta ad un lento sollevamento eustatico. La velocità di sollevamento è compresa tra il mm/a della costa occidentale e i 4 mm/a della costa orientale. I depositi glaciali flandriani nell’Olocene sono stati rimaneggiati dalle acque superficiali, ma con scarsa energia, sia a causa della modesta acclività dei rilievi, che della modesta entità del sollevamento tettonico. La carta pedologica del Regno Unito classifica i suoli della Great Valley come Gleysol.
Il Clima della Scozia è caratterizzato da intense precipitazioni, comprese tra 1000 e 2000 mm/a, omogeneamente distribuite nel corso dell’anno. L’evapotraspirazione nella contea di Cliland supera le precipitazioni tra aprile e settembre per un massimo di 40 mm (figura F311-1). Le temperature medie mensili sono comprese tra i valori minimi di 3-6 °C ed i valori massimi di 17-22°C. A causa della calda Corrente del Golfo che affiora a poche centinaia di Km a nord delle coste scozzesi i periodi di gelo e di copertura nevosa del suolo sono limitati a pochi giorni all’anno.
La contea di Cliland è ubicata a qualche decina di km a sud di Glasgow, la città che fino all’inizio di questo secolo rappresentava il principale centro industriale dell’Impero Britannico. Nella contea di Cliland l’attività economica prevalente è stata quella agricola fino al ‘700, epoca nella quale incomincia l’estrazione dalle rocce paleozoiche. All’attività mineraria si aggiunge all’inizio del ‘900 l’attività siderurgica dello stabilimento British Stell. Agli inizi degli anni ‘80 cessa l’attività mineraria e, poco dopo, nel 1987, quella siderurgica. Alla rilocalizzazione dell’industria pesante nei paesi in via di sviluppo nell’area metropolitana di Glasgow si accompagna l’espansione delle attività terziarie e l’urbanizzazione delle aree agricole e delle aree industriali dismesse. Il cambiamento d’uso del suolo suscita preoccupazione presso le autorità locali dell’area metropolitana di Glasgow a causa della presenza nel suolo di metalli pesanti nei suoli e del possibile effetto sanitario sulle popolazioni residenti.
Per valutare il grado di contaminazione del suolo della contea di Cliland si è campionato un profilo dello spessore di 45 cm a cinque Km sottovento lo stabilimento siderurgico della British Stell, sulla sommità pianeggiante di un Drumlins. Il suolo campionato nel corso del ‘900 a ricevuto le emissioni fumose dello stabilimento siderurgico della British Stell. Il flusso inquinante è stato confinato
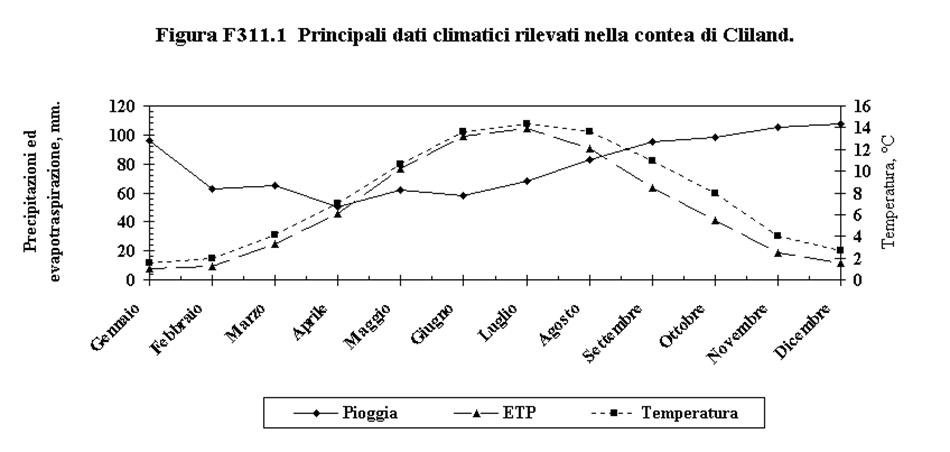
Figura F311-1. Dati climatici della
contea di Cliland.
nell’orizzonte Ap dalle lavorazioni agricole fino al 1987, anno in cui la British Stell ha smantellato lo stabilimento. Nello stesso anno il suolo è stato riconvertito dalla coltivazione dell’orzo a quella del prato permanente e sono quindi cessate le lavorazioni agricole. L’inquinante confinato nell’orizzonte lavorato ha così potuto essere lisciviato verso gli orizzonti profondi.
3.1.2. Il Comune di
Villadossola, Verbania, Italia.
L’area di studio si estende sottovento allo stabilimento siderurgico SISMA di Villadossola (Vb) su un’area di 20 Km2 circa, due chilometri ortogonalmente all’asse vallivo e cinque longitudinalmente (figura F312-1).
Il settore di valle studiato è intagliato in 5 diverse unità tettoniche (Barbieri F., 1983): la Falda Ofiolitica Piemontese, il Penninico, il Basamento Penninico, l’Austroalpino ed il Basamento Sdalpino. Lo schema tettonico dell’area di studio, e i rapporti strutturali tra le unità tettoniche sonno riportati nelle figure F312-2 ed F312-3. Le rocce presenti nell’alta Val d’Ossola (figura F312-3) sono principalmente Gneiss, Micascisti, Metabasiti, Calcescisti e, secondariamente, Serpentiniti. In tutta la valle si osservano mineralizzazioni ad Arsenopirite aurifera, Calcopirite, e Niccolite di età mesoalpina, riportate nella figura F312-2 con i simboli As o Au, Cu e Ni rispettivamente. In corrispondenza dell’anticlinale delle valli Anzasca ed Antrona, dove un lembo di Metabasiti della Falda Ofiolitica Piemontese è pizzicato all’interno degli Gneiss del Basamento Pennidico (figura F312-3, profilo A’-B’), le mineralizzazioni di Arsenopirite aurifera sono costituite da sistemi di vene dello spessore di qualche metro e di diversi ettometri di lunghezza. Le mineralizzazioni della Val d’Ossola costituiscono il centro di alto grado di un campo di mineralizzazioni a solfuri ellissoidale che si estende per qualche centinaio di Km parallelamente all’asse dell’edificio alpino Nord-Occidentale (Mastrangelo F., Natale P. e Zucchetti S., 1983), la cui origine va ricercata nella circolazione idrotermale associata all’attività plutonica delle radici dell’orogeno alpino.
Le mineralizzazioni della Val d’Ossola sono state coltivate per l’estrazione di oro e ferro ad incominciare dal 1700. L’attività estrattiva è stata nell’ultimo secolo condotta su scala industriale raggiungendo la massima intensità sul finire dell’ultima guerra. Nel complesso essa ha portato all’escavazione di un sistema di gallerie che si estende svariate decine di chilometri. La presenza di giacimenti minerari economicamente interessanti è, come per le altre vallate, la ragione della vocazione siderurgica della valle d’Ossola, lungo il cui asse si contano 6 fonderie. La storia dell’attività siderurgica della valle è complessa e parzialmente ricostruibile attraverso l’intervista dei quadri tecnici degli stabilimenti e delle miniere. E’ ragionevole ritenere che la fusione dei minerali escavati dai campi minerari della Valle d’Ossola sia andata progressivamente aumentando di pari passo con l’attività
Figura F312-1. Area di campionamento
della Val d'Ossola.
Figura F312-2. Schema tettonico
dell'area di studio.
Figura F312-3. Carta geologica
compilativa dell'alta Val d'Ossola.
estrattiva nel corso del ‘900, raggiungendo i valori massimi sul finire dell’ultima guerra. Successivamente alla chiusura delle miniere, avvenuta negli anni ‘50, le fonderie devono essersi orientate alla fusione di rottami e materiale minerario proveniente da giacimenti esteri. La storia dell’inquinamento dei suoli di Villadossola ascrivibile alle attività siderurgiche è presumibilmente complessa. L’evento inquinante di maggiore rilievo degli ultimi cinquanta anni sembra comunque risalire al biennio 1981-1982. In tale biennio la popolazione ha lamentato presso le Unità Sanitarie Locali ingenti e preoccupanti emissioni da parte dello stabilimento SISMA, le cui osservazioni sono riportate nella tabella T312-1. E’ possibile stimare l’emissione di metalli pesanti complessiva avvenuta nel biennio 1982-1983 assumendo che le emissioni orarie registrate dalle UUSSLL siano durate un’ora per ciclo fusorio e che vi siano stati 9 cicli fusori al giorno.
Tabella T312-1. Emissioni in atmosfera della fonderia SISMA nel biennio 1980-1982.
|
|
|
|
|
Elemento |
Emissione oraria, Kg/h |
Emissione complessiva, t |
|
Zn |
45 |
296 |
|
Cr |
20 |
131 |
|
Fe |
5,34 |
35,1 |
|
Pb |
5,0 |
32,9 |
|
Ca |
5,0 |
32,9 |
|
Mn |
3,0 |
19,7 |
|
Cu |
0,20 |
1,31 |
|
Ni |
0,11 |
0,72 |
|
Cd |
0,10 |
0,66 |
Nell’ultima glaciazione del Pleistocene, terminata circa 10.000 anni fa, la valle d’Ossola era coperta da un ghiacciaio della potenza di 1-2 Km. Sui versanti vallivi si rinvengono le tracce sedimentarie dell’episodio glaciale, rappresentate da morene laterali, depositi fluvio-glaciali ed infine da depositi loessici. Nel successivo periodo caldo olocenico, ovvero nel corso degli ultimi 10.000 anni, i depositi glaciali e periglaciali sono stati rimodellati dallo scorrimento delle acque superficiali. I depositi glaciali risultano così ora rimodellati dall’acqua non incanalata e dissecati dai corsi d’acqua. Essi possono essere osservati principalmente in corrispondenza della rottura di pendio associata alle spalle glaciali, in quanto lungo i versanti vallivi, se presenti, sono ricoperti da uno strato metrico di colluvium ridistribuito dallo scorrimento delle acque non incanalate. Nel fondovalle i depositi glaciali,
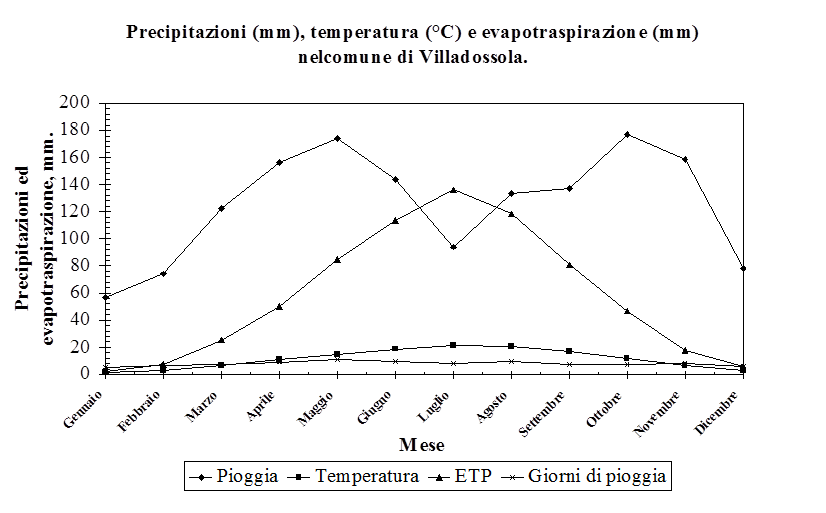
.
quando presenti, sono ricoperti dalle alluvioni deposte dal fiume Toce e dai suoi affluenti. Lo spessore del deposito alluvionale si può stimare raggiunga in corrispondenza dell’asse vallivo la potenza di 100-200 metri. Le alluvioni che occupano il fondovalle sono costituite da conglomerati e ghiaie deposti ad opera del fiume Toce e nei suoi affluenti. In superficie i conglomerati e le ghiaie sono ricoperte da uno strato dello spessore di qualche decimetro di sabbie fini e limi.. I depositi alluvionali del fondovalle sono rimaneggiati dalle acque superficiali con relativa frequenza rispetto ai depositi colluviali dei versanti vallivi.
La “Carta della capacità d’uso del suolo” edita dalla Regione Piemonte distingue Entisuoli sul fondovalle ed Inceptisuoli sui versanti vallivi. L’uso del suolo è in stretta relazione alla topografia. Nel fondovalle, pianeggiante, i suoli sono coltivati a mais in rotazione con prati polifiti e sono soggetti a lavorazioni meccanizzate relativamente profonde (20-30 cm). La profondità di aratura è limitata dalla presenza a modeste profondità di ghiaie e ciottoli. I suoli impostati sui versanti vallivi per essere coltivati sono stati terrazzati. A causa dell’idoneo microclima i terrazzamenti in passato hanno ospitato coltura della vite, mentre attualmente sono occupati prevalentemente da prati permanenti e a boschi cedui. La profondità di lavorazione sui terrazzamenti è limitata dal modesto spessore del deposito colluviale, dalle accentuate pendenze e dalle modeste dimensioni degli appezzamenti che rendono impossibile la lavorazione meccanica con mezzi pesanti. Le lavorazioni, quando vengono effettuate, sono leggere (10-20 cm). I suoli dei versanti vallivi che non sono stati terrazzati e non possono essere lavorati a causa dell’elevata acclività ospitano foreste di latifoglie o boschi cedui.
Il comune di Villadossola dal punto di vista climatico rientra nella Regione mesaxerica, sottoregione ipomesaxerica del sistema Bagnouls Gaussen (figura F312-4). La temperatura media annua è 11.5 °C, con le temperature maggiori corrispondenti ai mesi di Luglio e Agosto. Secondo i dati riportati dall'atlante climatologico della Regione Piemonte, subisce in media 64 giorni di gelo all'anno. La distribuzione della piovosità mostra 2 massimi annuali, uno in Maggio e l'altro in Ottobre con un totale di 1518 mm di precipitazioni all'anno. Il mese con il maggior numero di giorni piovosi è però Maggio. L'evapotraspirazione potenziale, secondo Thorntwaite, è invece massima durante i mesi di Giugno e Luglio. Secondo il Newhall Simulation Model, il regime termico dei suoli è mesico, mentre quello idrico è udico.
Per valutare lo stato di inquinamento dei suoli della contea di Cliland è stato raccolto un unico profili di suolo sulla sommità di un rilievo collinare alto un centinaio di m a 5 Km sottovento lo stabilimento siderurgico della British Steell. Il suolo è stato campionato raccogliendo una carota avente 10 dm2 di area e 45 cm di altezza. La carota è stata quindi suddivisa in 9 fette aventi 5 cm di spessore
Nel comune di Villadossola sono stati raccolti campioni in 19 siti in un’area che si estende per 5 km lungo l’asse vallivo e per 2 km ortogonalmente ad esso (F312-1). La densità di campionamento nel comune di Villadossola è stata di 1 sito per Km2, una densità di campionamento considerata più che sufficiente al fine della cartografia pedologica e ambientale. Nello scegliere l’ubicazione delle stazioni di campionamento si è cercato ovviamente di raccogliere un insieme rappresentativo e ragionevolmente bilanciato sia delle principali unità d’uso del suolo che delle principali unità geomorfologiche.
Nel confronto dell’uso del suolo si sono distinti i suoli coltivati a bosco, a prato permanente sfalciato e a prato sfalciato in rotazione agricola. Nei confronti della geomorfologia dell’area si sono distinti i suoli del fondovalle alluvionale, la cui roccia madre è rappresentata da depositi alluvionali, i suoli dei versanti vallivi, la cui roccia madre e il colluvium, e i suoli ubicati sulle spalle glaciali, sorta di pianori scolpiti a mezzacosta dalle lingue dei ghiacciai che nel pleistocene si insinuavano nella valle. La roccia madre dei suoli impostati sulle spalle glaciali è rappresentata da una sequenza sedimentaria costituita alla base da depositi fluvio-glaciali ed alla sommità dal colluvium.
Tenendo presente la geomorfologia e dell’uso del suolo nell’area si sono identificate sei diverse unità. In funzione della geomorfologia si sono distinte tre unità:
1) i versanti vallivi;
2) le spalle glaciali;
3) il fondovalle alluvionale;
Altre tre unità si sono distinte in ragione dell’uso del suolo:
1) forestale;
2) prato permanente;
3)
prato in rotazione agricola con colture
cerealicole;
Al fine di studiare la lisciviazione degli inquinanti si è considerata la storia recente dei suoli, e si sono distinti i suoli “disturbati”, il cui profilo è stato perturbato da lavorazioni successivamente all’evento inquinante del biennio 1981-1982 e suoli “indisturbati”.
I suoli sono stati campionati scavando una buca pedologica e suddividendo il profilo del suolo in fette prismatiche con area di 1 dm2 e dello spessore di 5 cm.. Il numero di strati raccolto attraverso il profilo varia da stazione a stazione ed è compreso tra il valore minimo di tre ed il valore massimo di sette. La profondità massima di campionamento è così compresa tra 15 e 35 cm. Sono stati inoltre campionati tre profili pedologici. In questi il profilo è stato diviso in orizzonti pedologici omogenei, che sono stati descritti secondo le norme della Soil Taxonomy; di questi è stato raccolto un volume di campione rappresentativo dell’orizzonte pedologico. Per la determinazione della densità apparente sono stati raccolti opportuni campioni mediante un cilindro in ottone avente lunghezza di cm 5 e diametro di 5, per un volume totale di circa 100 cm3.
I campioni di suolo sono stati asciugati a temperatura ambiente. L’umidità relativa e la densità apparente sono stati determinati sui campioni raccolti con il carotatore cilindrico per differenza tra il peso secco ed il peso umido. Le analisi chimico-fisiche sono state effettuate sulla terra fine (<2 mm), separata dallo scheletro con setaccio di nylon. I risultati ottenuti sono stati quindi riferiti ad un kg di terra fine oppure ad un dm3 di suolo.
La porosità apparente (g·dm-3) è stata determinata con il metodo del carotatore cilindrico (Società Italiana della Scienza del Suolo, 1985). Per ogni strato di suolo dello spessore di 5 cm si sono prelevati due cilindri dell’altezza di 5 cm e di 5 cm di diametro, raccogliendo un totale di circa 200 cm3 di suolo per strato. La porosità apparente è stata calcolata quale rapporto del peso del suolo seccato a temperatura ambiente ed il volume campionato.
La tessitura è stata determinata gravimetricamente per levigazione alla pipetta con l’apparecchio di Gattorta previa dispersione con Na-esametafosfato. 10 g di terra fine sono stati posti in bottiglia di polivinile, umettati con una soluzione di 10 ml di sodio esametafosfato 11,7% e dispersi in 100 ml di acqua deionizzata. La bottiglia di polivinile è stata posta ad agitare per 60’. La sabbia grossa (2 mm<Ø) è stata separata per setacciatura ad umido. La sospensione contenente sabbia fine, limo ed argilla è stata posta in un levigatore di Gattorta avente 500 ml di capacità e 50 cm di altezza. Ad intervalli di tempo calcolati secondo la legge di Stock si sono effettuati due prelievi di 10 ml di sospensione contenenti rispettivamente le frazioni tessiturali limo (0,02 mm < Ø < 0,002 mm) + argilla (Ø<0,002mm) ed argilla. La sospensione prelevata è stata posta in un vetrino da orologio, seccata a 60°C e pesata con la bilancia di precisione in atmosfera mantenuta anidra con gel di silice. La percentuale di argilla, limo, e sabbia grossa è stata calcolata direttamente del peso del sedimento prelevato, mentre la percentuale di sabbia fine è stata calcolata come complemento a cento. Nella determinazione del contenuto di argilla si è considerata la densità della colonna d’acqua nella quale è avvenuta la sedimentazione e la presenza del sodio esametafosfato impiegato come disperdente, variabili entrambe non trascurabili.
La sostanza organica è stata determinata mediante ossidazione ad umido con potassio dicromato secondo il metodo Walkley-Black seguendo i metodi normalizzati della S.I.S.S. (Società Italiana della Scienza del Suolo, 1985). 10 g di terra fine sono stati macinati con un mortaio di agata fino ad ottenere un macinato avente dimensione inferiore a 0,5 mm. Una aliquota di macinato compresa tra 0,5 e 2,0 g è stata posta in un matraccio da 250 ml e messa a contatto con 10 ml potassio dicromato 0,100 N. La reazione di ossidazione della sostanza organica è stata innescata dall’aggiunta di 20 ml di acido solforico concentrato ed interrotta dopo 30’ esatti dall’aggiunta di 200 ml di acqua deionizzata. Nel corso della reazione di ossidazione il matraccio è stato mantenuto in agitazione per garantire una omogenea reazione del campione di suolo. Il dicromato ridotto a cromito dalla reazione con la sostanza organica è stato determinato per differenza titolando il dicromato residuo con il sale di Mohor, Fe(NH4)2(SO4)2, 0,050 N. Come indicatore della titolazione si è impiegata ferroina. La quantità di suolo sottoposta ad ossidazione è stata opportunamente pesata così che avere sempre almeno 0,15 mEq di dicromato in eccesso rispetto alla sostanza organica.
La capacità di scambio cationico (CSC) è stata determinata mediante scambio con bario cloruro BaCl2 tamponato a pH 8,15 con trietanolammina seguendo i metodi normalizzati della S.I.S.S. (Società Italiana della Scienza del Suolo, 1985). 2,0 g di terra fine macinata a 0,5 mm sono stati posti in un tubo da centrifuga con 25 ml di bariocloruro BaCl2 al 10% tamponato a pH 8,15. Il suolo ed il reagente si sono mantenuti in agitazione per 20’ così da ottenere un completa reazione del Ba con il complesso di scambio. Il Ba+2 non adsorbito dal suolo è stato rimosso con due successivi lavaggi effettuati con 25 ml di acqua deionizzata intercalati da 10’ di agitazione meccanica. Al suolo saturato in Ba+2 e liberato dall’acqua deionizzata di lavaggio mediante centrifugazione si sono aggiunti 25,0 ml di MgSO4 0,100 N. Lo scambio tra il Ba+2 adsorbito sul suolo ed il Mg+2 è stato garantito dall’agitazione della sospensione per 20’ e dalla precipitazione del BaSO4. La quantità di Mg+2 adsorbita sul complesso di scambio è stata determinata per differenza titolando con EDTA il Mg+2 rimasto in soluzione dopo lo scambio con il Ba.
Il pH è stato determinato potenziometricamente in acqua ed in KCl 1N secondo i metodi normalizzati di analisi del suolo della S.I.S.S. (op. cit.). 2 g di terra fine sono stati posti in bottiglia di polietilene e messi a reagire con 5 ml di acqua deionizzata. Il suolo è stato lasciato reagire con l’acqua deionizzata per 15’ su agitatore meccanico, quindi lasciato sedimentare per 30’. L’elettrodo del pH-metro è stato immerso nel liquido limpido e la lettura effettuata dopo 5’.
L’elemento “totale” è stato determinato mediante estrazione con acqua regia in canne a riflusso secondo i metodi normalizzati della S.I.S.S. (op. cit.). 1 g di terra fine macinata a 0,5 mm è stata posta in una canna a riflusso ed addizionata con 7,5 ml di acido cloridrico concentrato e 2,5 ml di acido nitrico concentrato. Le canne a riflusso sono state poste a bollire su un bagno di sabbia per due ore. Una volta raffreddata, la sospensione contenuta nelle canne a ricadere è stata centrifugata, il residuo solido lavato con acqua deionizzata e il surnatante portato ad un volume finale di 100 ml esatti.
L’elemento biodisponibile è stato determinato mediante estrazione con il reagente di Lakanen (una soluzione di ammonio acetato 0,5 M + EDTA 0,02 M tamponata a pH 4,65) secondo i metodi normalizzati di analisi del suolo della S.I.S.S. (op. cit.). 10 g di terra fine (Ø<2 mm) sono stati posti in bottiglia di polietilene e messi a reagire con 100 ml del reagente di Lakanen su agitatore meccanico per 60’. Il surnatante è stato separato per centrifugazione e filtrazione.
Le concentrazioni di Si, Fe, Al, Ca, Mg, K, P, S, Cr, Mn, Co, Ni, Zn e Cu, nell’acquaregia è stata effettuata direttamente sull’estratto mediante ICP Perkin-Elmer Optima 3000 multicanale, con rilevatore echelle. Le concentrazione di Bi, As ed Sb nell’acquaregia sono state misurate mediante ICP Perkin-Elmer Optima 3000 multicanale previa generazione e concentrazione degli idruri secondo il metodo e le apparecchiature sviluppate da Morrow A., Wiltshire G., e Hursthouse A., (1997). Dell’analita 10 ml sono stati posti in un matraccio da 25 ml ed addizionati di 75 mg di ioduro di potassio, 75 mg di acido ascorbico e 3 ml di acido cloridrico concentrato. La soluzione è stata lasciata a riposo per 20’, quindi portata a volume con acqua deionizzata. Il preparato è stato fatto reagire per qualche secondo con una soluzione di sodio boroidrato NaBH4 1 N mantenuta a pH 12 con idrossido di sodio mediante un’apposita apparecchiatura costituita da una pompa peristaltica accoppiata ad un anello miscelatore. Gli idruri di As, Bi ed Sb generati dalla reazione dell’analita con il sodio boroidrato sono stati quindi raccolti dall’anello miscelatore mediante un separatore a membrana ed aspirati dalla torcia al plasma dell’ICP. La concentrazione di Cd e Pb nell’acquaregia è stata misurata mediante fornetto di grafite Perkin-Elmer HGA 700. All’analita è stata aggiunto un condizionatore di matrice e si sono effettuate le correzioni dell’interferenza della matrice mediante lampada a raggi ultravioletti. Le concentrazioni di Co, Ni, Zn, Cu, Cd e Pb nella soluzione estratta dal reagente di Lakanen è stata determinata mediante assorbimento atomico.
La mineralogia della frazione argillosa è stata determinata con spettrofotometro a polveri Siemes modello D5000 impiegando la radiazione CuK, una fenditura Soller sul raggio primario e un monocromatore a grafite sul raggio secondario. I trattamenti impiegati per la determinazione semiquantitativa sono stati quelli indicati da Thorez J., (1976) e Wilson M.J. (1987):
1) campione saturato in Mg;
2)
campione saturato in K;
2) saturato con glicole etilenico;
3) riscaldato a 550°C.
La composizione mineralogica è stata quantificata numericamente assumendo che l’altezza del picco diagnostico del minerale considerato sia direttamente proporzionale alla sua percentuale in peso. La percentuale di Illite è stata così stimata dall’altezza del picco a 10 Å del campione saturato in Mg, la percentuale di Vermiculite è stata calcolata dall’abbassamento del picco a 14Å a seguito della saturazione del campione con K e La percentuale di Smectite è stata calcolata dall’altezza del picco a 16 Å del campione trattato con glicole etilenico. La percentuale di Caolino, infine, è stata calcolata dall’abbassamento del picco a 7 Å a seguito del riscaldamento a 550°C.
Il metodo adottato trascura i fattori di struttura, la cristallinità e l’effetto dell’interstratificazione dei minerali sull’altezza e la forma del picco di diffrazione del minerale considerato ed è pertanto semiquantitativo (Wilson M.J., 1987). A causa dei fattori di struttura il metodo sottostima sistematicamente la percentuale in peso dei minerali poveri di metalli pesanti e con strutture cristalline disordinate mentre sovrastima i minerali ricchi di elementi pesanti e con strutture cristalline ordinate. Il fenomeno dell’interstratificazioni dei minerali argillosi ha effetto sulla ampiezza e sulla forma del picco di diffrazione e dipende dall’ordine con cui si susseguono i foglietti dei diversi minerali. Trascurare il fenomeno dell’interstratificazione dei minerali argillosi determina una sottostima o una sovrastima dei minerali in dipendenza dell’abbondanza relativa dei minerali nel cristallo e dall’ordine con cui si susseguono nell’edificio cristallino.
La mineralogia della frazione sabbiosa e limosa è stata determinata mediante microsonda elettronica a dispersione di energia SEM-EDS. Le zollette di suolo analizzate sono state inglobate in resina epossidica, quindi sezionate e lucidate. La sonda è stata calibrata sull’emissione K del Co. Per la quantificazione degli elementi primari si sono impiegate rette di taratura calibrate su minerali standard. La determinazione quantitativa così effettuata è affetta dall’errore determinato dalla differenze di densità dei minerali analizzati e di quelli impiegati nella retta di taratura dello strumento. Tali errori sono trascurabili ai fini della presente indagine.
Le analisi micromorfologiche sono state effettuate alla microsonda elettronica SEM. Le zollette di suolo sono state poste sul piattello portacampioni e ricoperte da un sottile strato di carbonio.
Validazione dei dati analitici. Le principali caratterizzazioni chimico-fisiche dei suoli (tessitura, contenuto in sostanza organica, CSC, pH, elemento estraibile dal reagente di Lakanen ed in acquaregia) sono state effettuate in doppio. La deviazione standard delle misure effettuate al di sopra dei limiti di rilevabilità metodologica e strumentale è risultata sempre inferiore al 10%. I limiti di rilevabilità strumentale e metodologica sono stati ampiamente superati per tutte le proprietà misurate eccetto che il contenuto di argille nei suoli di Villadossola, dove talvolta si osserva tra i replicati una deviazione standard del 30%.
Nelle misure effettuate all’ICP di ogni analita si sono considerate le emissioni determinate dalla matrice ed effettuata l’adeguata sottrazione della radiazione di fondo. Per tutti gli elementi le concentrazioni sono state misurando due diverse linee di emissione, tra le quali hanno si è sempre osservato un coefficiente di varianza inferiore al 3%. Le deviazioni standard tra i replicati, eccetto che per As e Bi, dove la deviazione standard si è mantenuta nel limite di incertezza metodologica del 30% , sono sempre state inferiori al 10%. Nella determinazione del Cd all’HGA, pur essendo stata effettuata ben al di sopra dei limiti di rilevabilità strumentale, la deviazione standard tra i replicati ha raggiunto valori medi del 20% con punte del 40%. Difficoltà nella determinazione del Cd all’HGA sono state lamentate dai tecnici che erano, indipendentemente, impegnati in analisi di campioni simili in laboratori sia italiani, presso il Dipartimento di Chimica Analitica e Strumentale della Università di Torino, che scozzesi, presso il Department of Chemistry and Chemical Engineering dell’Università di Paisley, Scozia. Le ragioni della elevata deviazione standard osservata tra i replicati delle misure effettuate all’HGA non è stata appurata, ma potrebbe essere dovuta a contaminazioni accidentali determinate dal campionatore dello strumento.
Gli strumenti statistici impiegati nella interpretazione dei risultati sono stati le correlazioni lineari e le regressioni multiple (Busetti G., 1983. Spiegel M.R., 1979). Le regressioni multiple sono state ottenute con il metodo dell’aggiunta progressiva di variabili. Una prima serie di regressioni sono state effettuate per ogni variabile dipendente considerata singolarmente. Quindi è stata selezionata la regressione con coefficiente di correlazione r2 più alto e limite di confidenza P pù significativo. Alla correlazione lineare così ottenuta sono state aggiunte una alla volta le rimanenti variabili. Delle numerose regressioni multiple così ottenute si è selezionata quella in cui entrambe le e la variabili indipendenti sono risultate significativamente correlate alla variabile dipendente e con maggior limite di confidenza. Si è quindi proceduto con metodo euristico aggiungendo progressivamente una alla volta tutte le altre variabili fino ad identificare tutte le proprietà del suolo significativamente correlate alla variabile dipendente.
La determinazione della concentrazione dell’elemento litogenico ed antropico di un campione di suolo riveste notevole importanza scientifica e normativa. E’ ovvio infatti che piccoli errori nella valutazione della concentrazione litogenica di un elemento possono portare a grandi errori nella stima del fattore di arricchimento antropico ovvero della perturbazione dei cicli biogeochimici naturali. E’ altrettanto ovvio che la distribuzione degli elementi nei minerali, nelle rocce e nei suoli non a causa dei processi geologici è disomogenea. Si possono così osservare in prossimità di particolari formazioni geologiche, quali i giacimenti minerali, elevate concentrazioni di elementi pesanti tossici la cui componente antropica è trascurabile anche se è, in termini di impatto sanitario, massiccia.
In letteratura sono stati riportati diversi metodi per la determinazione della concentrazione dell’elemento litogenico ed antropico (Adriano D.C., 1986. Alloway B.J., 1997. Salomons W. e Förstner U., 1984), nessuno dei quali sembra essere completamente soddisfacente od universalmente accettato. Nei seguenti paragrafi si discutono le caratteristiche dei metodi riportati in letteratura e di quelli adottati nella presente ricerca.
Quando non si possa disporre di costose misure isotopiche o quando la composizione isotopica dell’inquinante e del suolo non differiscono sufficientemente, la misurazione diretta della concentrazione litogenica non è possibile e si ricorre pertanto ad assunzioni e stime ragionevoli.
Alcuni autori, assumono che la concentrazione litogenica dei suoli sia quella degli strati ubicati a profondità superiori a 40 cm. Altri autori prendono come riferimento la concentrazioni degli elementi in profili pedologici raccolti lontano dalla sorgente di inquinamento.
Entrambe i metodi presentano alcuni limiti. I processi pedogenetici infatti comportano una ridistribuzione degli elementi attraverso il profilo di suolo e conducono alla formazione di orizzonti differenti per proprietà fisiche e chimiche (Sequi P. Ed, 1986. Vinogradov A. P., 1959). Gli orizzonti superficiali di suolo così, anche quando non contaminati, hanno una composizione chimica differente da quelli profondi. Tra i processi che possono portare ad un accumulo di un elemento negli orizzonti superficiali di suolo (Violante P., 1986b) vi è ovviamente la precipitazione dell’elemento liberato nel corso dell’alterazione pedogenetica dei minerali primari in forme immobili quali quelle occluse nei reticoli dei minerali secondari, quali argille, sesquiossidi, idrossidi, fosfati, solfati, o carbonati. Inoltre, per gli elementi che possono essere bioaccumulati nella biomassa vegetale quali Cu, Cd, As, Bi, l’elemento è trattenuto negli orizzonti superficiali ricchi in sostanza organica (Whedepohl K. H., 1969. Vinogradov A. P., 1959). Tra i processi che comportano l’impoverimento di un elemento negli strati superficiali di suolo vi è la traslocazione preferenziale sotto forma di composti solubili facilmente lisciviabili quali PbCl3-1, Cu(OH)3-1 , CrO-, Zn(OC-CH4)3-2, Co(HSO4)3-1 Pb+2, PbCl20, o complessi organometallici con molecole organiche solubili a basso peso molecolare (Cu-OOC-CH3). Inoltre semplici modelli cromatografici che simulano la lisciviazione degli inquinanti poco mobili (Konshin O. V., 1982), indicano che quando la velocità di lisciviazione dell’inquinante è sufficientemente elevata, la concentrazione dell’inquinante antropico negli strati profondi può non essere trascurabile. Il 137Cs, per esempio, può essere in taluni suoli lisciviato alla velocità media di 1 cm/a (op. cit.), venendo così a trovarsi dopo soli 50 anni dalla contaminazione in concentrazioni apprezzabili alla profondità di 1 m.
L’identificazione
di suoli incontaminati simili a quelli in oggetto di studio può sembrare il
metodo migliore per determinare la distribuzione litogenica di un elemento
attraverso il profilo del suolo, in quanto il metodo tiene conto dei processi
naturali che ne determinano l'ineguale distribuzione attraverso il profilo.
Recenti studi hanno messo in luce che il
metodo è più difficoltoso di quanto in prima analisi si potrebbe essere indotti
a ritenere e può per alcuni elementi risultare assai fuorviante. Per il Pb, per
esempio, la cui composizione isotopica permette di scorporare l’elemento
litogenetico da quello antropico, sono state registrate concentrazioni di
elemento antropico di alcuni ordini di grandezza superiori al contenuto
litogenetico anche in aree lontane centinaia di km dalla più vicina sorgente di
inquinamento (Shirata H., Elias R.W. e Patterson C. C., 1980. Alloway B.J.,
1997. Salomons W. e Förstner U., 1984). Le elevate deposizioni atmosferiche
registrate in aree lontane centinaia di km da sorgenti inquinanti, i
rilevamenti condotti nelle campagne dei paesi industrializzati (Bertelsen B.O.,
et al. 1995, Davies B.E., 1997) e il contenuto di Pb presente nelle carote di
ghiaccio artico suggeriscono che la maggior parte del Pb misurato nei suoli
dell’emisfero nord sia di origine antropica. Ipotesi che sembra confermata
dall’osservazione che il fattore di arricchimento del Pb nel suolo rispetto
alla roccia madre osservata mostra un progressivo aumento nel corso di questo
secolo da 0,8 in Russia nella prima metà del ‘900 (Vinogradov A. P., 1959) a
1.4 negli anni ‘80 nel Nord America (Shacklette H. T. e Boerngen J. G.).
Elevate concentrazioni di Pb antropico si osservano anche in carote provenienti
da bacini lacustri, la cui datazione indica che la deposizione del Pb al suolo
ha inizio nella prima metà del ‘700 in Inghilterra (Farmer J. G. e Eades L.J.,
1996) e nella seconda metà dell’800 in Nord America (Shirata H., Elias R.W. e
Patterson C. C., 1980), presumibilmente sia a causa della combustione del
carbone, che al progressivo aumento delle attività siderurgiche connesse
all’industrializzazione.. L’inquinamento del suolo da Pb è pertanto un fenomeno
che interessa quantomeno tutto l’emisfero settentrionale, e in nessun’area dei
paesi industrializzati dell’emisfero nord, per quanto remota, è quindi possibile
rinvenire suoli che non siano sensibilmente contaminati dai quali poter quindi
ricavare la concentrazione litogenica dell’elemento attraverso il profilo. Per
un numero indefinito di elementi, quali Cd, Zn, Cu, As, Bi, Sb che sono associati ai solfuri e che come il
Pb sono mobilizzati nel corso dell’estrazione, delle attività siderurgiche,
industriali ed agricole in quantità complessive comparabili a quelle
mobilizzate dai processi geologici, è quantomeno plausibile attendere una
analoga contaminazione globale, tale da rendere vana la ricerca di profili di
suolo sicuramente incontaminati.
Nella presente ricerca la determinazione della concentrazione litogenica degli elementi in traccia è stata determinata mediante un modello geochimico. Il modello adottato si basa su 4 assunti:
1) il
sedimento morenico sul quale si sviluppano i suoli studiati è costituito da un
miscuglio omogeneo di detriti litoidi e minerali ablasi dalle rocce che
affiorano nell’area coperta dal ghiacciaio che ha deposto la morena;
2) la
concentrazione dell’elemento in traccia nelle rocce nell’area di studio è
normale, ovvero uguale alla composizione media di rocce simili raccolte in
tutto il mondo;
3) il
contributo di un litotipo affiorante nell’area interessata dall’esarazione
glaciale nel determinare la composizione della morena è direttamente
proporzionale alla relativa superficie di affioramento;
4) i processi pedogenetici in atto nell’area di studio non hanno determinato né un arricchimento né un impoverimento dell’elemento nel profilo dei suoli.
Seguendo questi assunti la concentrazione dell’elemento in traccia nel suolo è stata quindi stimata uguale a alla concentrazione media dell’elemento nelle affioranti nell’area ponderata per la rispettiva superficie di affioramento.
Il modello adottato per stimare la concentrazione litogenetica dei suoli di Villadossola è uguale a quello impiegato per la determinazione della composizione della crosta superiore, essendo questa supposta essere uguale alla composizione delle morene deposte dai ghiacciai continentali, che in quanto tali, hanno ablaso e miscelato un campione rappresentativo delle rocce della crosta superiore. Questa stima fornisce in verità risultati in buon accordo con quelle effettuate per vie indipendenti dimostrandosi sufficientemente accurata per gli scopi proposti (D’Amico C. et al., 1989). L’applicazione del modello geochimico proposto trova ovviamente la ragione d’essere nella origine dei sedimenti sui quali si sviluppano i suoli. Questi sono infatti di origine glaciale e sono il frutto dell’ablazione e del trasporto su distanze di chilometri (Villadossola) delle particelle distaccate dalla roccia madre ad opera dei ghiacciai che nel Pleistocene inferiore coprivano il bacino di afferenza.
Il
metodo di stima adottato presenta tuttavia alcuni limiti. Innanzi tutto
trascura la azione di differenziazione del sedimento di origine glaciale
operata dallo scorrimento delle acque superficiali. Nel corso dell’Olocene le
acque superficiali hanno infatti agito ridistribuendo sulla superficie
topografica i granuli minerali presenti nel suolo in funzione del loro
diametro, della densità e della pendenza della superficie topografica. È per
contro ragionevole ritenere che l’effetto del rimodellamento superficiale delle
acque, più che spostare il valore medio osservato dal valore della
concentrazione litogenica vera, abbia l’effetto di distribuire la
concentrazione litogenica dell’elemento attorno al suo valore medio originario.
Come già visto, il modello trascura inoltre l’effetto che i processi
pedogenetici hanno nel ridistribuire gli elementi in modo difforme attraverso
gli orizzonti del suolo.
Il modello adottato presenta infine il limite di essere valido a descrivere la composizione della roccia madre quale è determinata da una digestione completa del suolo. Nella determinazione del contenuto totale degli elementi in traccia considerati si è impiegata al contrario l’acquaregia. L’acquaregia dissolve la sostanza organica, gli idrossidi, i carbonati, i solfati, fosfati, i solfuri, i serpentini e solo parzialmente le argille e i Fillosilicati (Page A.L., Miller R.H., e Keney D.R. Eds, 1982). L’elemento occluso nella maggior parte dei minerali primari silicatici e in alcuni spinelli primari non è dissolta dall’attacco in acquaregia. E’ pertanto lecito concludere che il modello geochimico adottato per determinare il contenuto litogenetico dei metalli pesanti comporti per quegli elementi che sono inclusi nei reticoli dei minerali inattaccabili all’acqua regia una sottostima della concentrazione litogenica.
La determinazione antropica dell’elemento dovrebbe essere calcolata come semplice differenza tra l’elemento totale e quello litogenico. In realtà, come discusso nel paragrafo precedente, la determinazione della concentrazione litogenica dell’elemento estraibile in acquaregia è difficoltosa e risente molto della composizione mineralogica della roccia madre e dell’evoluzione pedogenetica del suolo (Whedepohl K.H., 1969. Vinogradov A.P., 1959. Sequi P., Ed., 1986). La valutazione della concentrazione antropica dell’elemento è così stata effettuata con due metodi diversi negli Entisuoli della Valle d’Ossola e nel Glaysol della Scozia.
Nel Glaysol di Cliland, fortemente lisciviato, è stato assunto seguendo Page et al. (1982) che l’elemento estraibile dall’acquaregia costituisca prevalentemente l’elemento antropico, mentre l’elemento litogenico sia per lo più occluso nei minerali silicatici non dissolti dall’acquaregia.
Nei suoli di Villadossola, che si sviluppano sui sedimenti di un bacino imbrifero sede di un corposo campo minerario, la concentrazione antropica è stata stimata essere uguale alla differenza tra la concentrazione totale ed il valore minore tra la concentrazione litogenica stimata con il modello geochimico e la concentrazione osservata nello strato più profondo di suolo. Il metodo di stima adottato per i suoli di Villadossola trova la sua giustificazione nell’osservazione che la concentrazione totale degli inquinanti nei suoli non disturbati dalle lavorazioni agricole tende a diminuire con la profondità fino e ad approcciare a profondità superiori ai 20 cm la concentrazione litogenica stimata.
Come è noto, la lisciviazione degli inquinanti attraverso la zona non satura può essere descritta da una numerosa gamma di modelli sia fisici che matematici (Feddes R.A. et al., 1988). Prima di esporre il modello adottato nella presente ricerca e discuterne le caratteristiche è pertanto opportuno valutare criticamente le caratteristiche dei principali modelli riportati in letteratura.
La lisciviazione degli inquinanti attraverso la zona non satura può essere descritta da una famiglia di modelli fisici nota con il nome collettivo di modelli “cromatografici advettivo-diffusivi”. Questi modelli nel descrivere la lisciviazione possono prendere in considerazione una vasta gamma di combinazioni di processi fisici fondamentali quali la diffusione, l’advezione, l’adsorbimento, la comparsa o la scomparsa per decadimento dell’inquinante, l’assimilazione da parte dei vegetali, le reazioni di complessazione ed adsorbimento che hanno luogo nella soluzione circolante. Questi modelli, in dipendenza della loro complessità, per descrivere la lisciviazione dell’inquinante necessitano la determinazione di numero variabile di proprietà del suolo e dell’elemento, quali la porosità, la permeabilità, la tortuosità del suolo, le isoterme di adsorbimento sui siti di scambio del mezzo poroso, le precipitazioni, l’infiltrazione efficace, l’evapotraspirazione, il coefficiente di diffusione dell’inquinante nella soluzione circolante, la composizione della fase fluida, la densità volumica delle radici, la traspirazione, nonché le leggi che regolano la comparsa o il decadimento dell’elemento.
Alcuni dei modelli cromatografici diffusivo-convettivi più sofisticati, quali è GEOCHEM (Lichtner P.C., 1988), non si limitano a considerare il sistema suolo-soluzione come un sistema chimico in stato di equilibrio, bensì lo considerano un sistema in equilibrio cinetico. Questi modelli considerano pertanto le cinetiche delle reazioni che in esso avvengono, tra le più importanti delle quali vi è la dissoluzione dei minerali primari, la precipitazione di minerali secondari o l’accumulo e l’ossidazione della sostanza organica. Come dimostrato da prove di terreno questi modelli descrivono efficacemente la migrazione degli elementi attraverso il suolo e le forme che assumono oltreché nei tempi di interesse prettamente pedologico anche su quelli geologici. Tali modelli, per quanto estremamente interessanti ed utili, sono estremamente complessi, e per essere applicati richiedono la conoscenza di tutte le reazioni che avvengono nel sistema considerato, delle costanti chimiche che le descrivono e di una approfondita caratterizzazione delle fasi che compongono il sistema studiato nelle sue condizioni iniziali. L’applicazione di questi modelli risulta così ardua, o, quando non si dispongono delle appropriate costanti chimico-fisiche, impossibile.
Esistono comunque modelli cromatografici convettivo-diffusivi assai più semplici e pur sempre efficaci a descrivere la lisciviazione degli inquinanti. Tra i più semplici modelli cromatografici advettivo-diffusivi applicati con successo in condizioni di terreno si cita quello sviluppato da Konshin O. V. (1982). Il modello di Konshin descrivere la lisciviazione degli inquinanti prendendo in considerazione i processi chimico-fisici dell’adsorbimento, dell’advezione, della diffusione e della dispersione. L’equazione differenziale che descrive la lisciviazione dell’inquinante è espressa dalla legge di Fick nello spazio monodimensionale:
dC(x,t)/dt = D·(d2C/dx2) - V·(dC/dt)
dove
V = velocità di lisciviazione dell’inquinante nel mezzo poroso quale determinata dalla percolazione delle acque e dall’adsorbimento dell’inquinante sulle superfici del mezzo poroso;
D = la costante che esprime i fenomeni della diffusione e della dispersione;
C(x,t) = concentrazione dell’inquinante al tempo t nel punto x;
La soluzione dell’equazione di Fick per le condizioni al contorno
(t=0 & x=0) C(0,0)=C0
trovata da Konshin è:
(C/C0)x,t = 1/[2·(·D·t)0,5] exp [-(x-V·t)2/(4·D·t)]
Il
modello di Konshin descrive la lisciviazione dell’inquinante attraverso il
suolo come una gaussiana che si sposta alla velocità V e la cui deviazione
standard aumenta alla velocità D·t (F371-1).
L’applicazione del modello di Konshin al suolo inquinato da 137Cs permette di osservare che la velocità V con cui l’inquinante è lisciviato non è costante attraverso il profilo, ma aumenta con la profondità (op. cit.). Mediante opportune interpolazioni è possibile trovare un’equazione che esprime la variazione di V e D con la profondità. A loro volta V e D possono essere messe in relazione alle proprietà chimico-fisiche del suolo, permettendo così di evincere i meccanismi ed i processi della lisciviazione. L’applicazione del modello di Konshin allo studio della lisciviazione degli inquinanti è uno strumento che fornisce utili informazioni. La sua applicazione alla determinazione delle proprietà
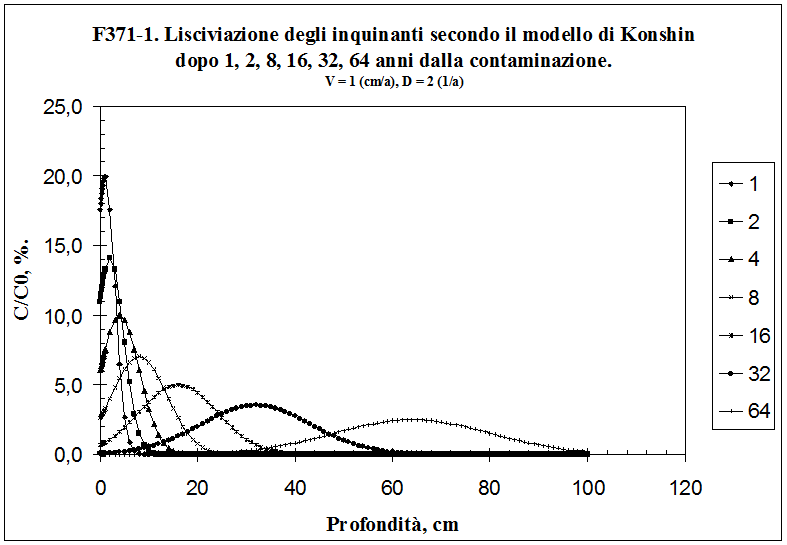
Figura 371-1. Lisciviazione degli
inquinanti secondo il modello di Konshin.
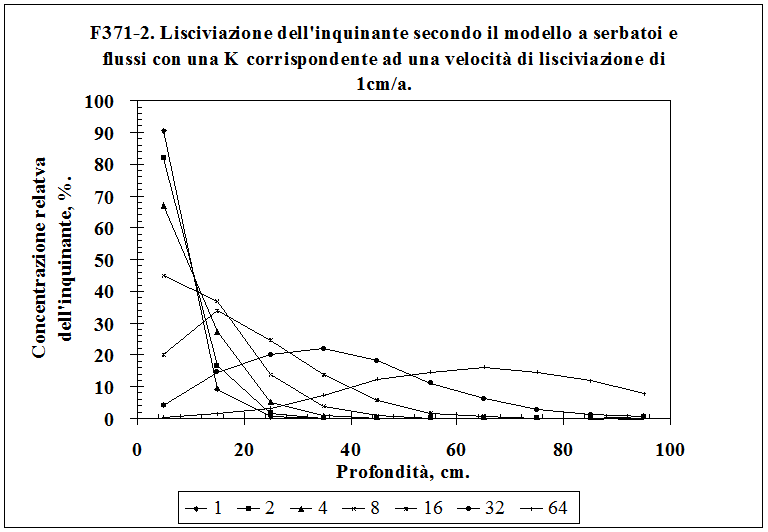
Figura 371-2. Lisciviazione degli
inquinanti secondo il modello di Ropolo, Bonazzola e Facchinelli.
del suolo che determinano la velocità di lisciviazione è comunque difficoltosa in quanto richiede lo sviluppo di un opportuno programma di calcolo che permettano di calcolare come V e D variano attraverso il profilo e come queste sono in relazione alle proprietà del suolo.
Recentemente per descrivere la mobilità degli inquinanti è stata proposta l’applicazione di modelli appartenenti alla famiglia dei modelli matematici a serbatoi e flussi. Tali modelli matematici hanno già trovato impiego nello studio della perturbazione antropica dei cicli biogeochimici (Lasaga A. C., 1980. Whitfield M, 1981. Lerman A. e Mackenzie F.T., 1975), nella valutazione dell’impatto sanitario determinato dalla contaminazione dei suoli (Sheppard S.C., 1995. Zach R. e Sheppard S. C., 1991) e della lisciviazione degli inquinanti (Poelstra P. e Frissel M.J., 1967. Bonazzola G. C., Ropolo R. e Facchinelli A., 1993).
Il modello a serbatoi e flussi impiegato da Bonazzola G. C. et al. (1993) descrive la lisciviazione dell’inquinante attraverso la determinazione empirica di una sola costante K caratteristica del profilo considerato (F371-2):
S(n,t) = e(-K·t) · (m=0m=n) S(m,0)·[k·t](n-m)·[(n-m)!]-1
Dove:
S(m,0) = la concentrazione dell’inquinante nello strato numero m al tempo t=0;
K = la costante cinetica del rilascio da un serbatoio al sottostante;
t = tempo intercorso dalla contaminazione.
. La costante empirica K che descrive la lisciviazione dell’inquinante attraverso il profilo è la cinetica del rilascio da uno strato di suolo al sottostante K che meglio interpola il profilo osservato. Il limite del modello adottato è che descrive il fenomeno della lisciviazione nei termini di una sola costante che riassume empiricamente i processi di advezione e dispersione, e non permette di determinare quali proprietà del suolo influiscono rispettivamente su questi due fenomeni. Infine il modello descrive la velocità di lisciviazione con costante cinetica del rilascio mediata sull’intero profilo e non permette pertanto di apprezzare come la velocità di lisciviazione varia di strato in strato nel singolo profilo e di trovarne le relazioni con le proprietà chimico-fisiche come il modello di Konshin rende possibile. Il vantaggio del modello a serbatoi e flussi rispetto al modello cromatografico advettivo-diffusivo di Konshin è che per descrivere la velocità di lisciviazione necessita del campionamento di un numero limitato di strati di suolo. Esso può essere applicato quando nel corso di
Figura F372-1. Il
modello a serbatoi e flussi ad n costanti.

un rilevamento di un’area inquinata si campionano per ogni suolo anche due strati soltanto, nel qual caso si determina la costante K del solo primo. Esso può quindi essere applicato anche nel corso di rilevamenti, quale quello presentato in questo studio, dove il numero di campioni da raccogliere è limitato dai costi economici e dev’essere distribuito su un’ampia superficie.
3.7.2. Sviluppo del modello.
Per sviluppare il modello si assume che il profilo del suolo sia assimilabile ad una serie di Sn strati, o serbatoi (F372-1), sufficientemente piccoli da poter essere considerati omogenei da un punto di vista delle proprietà chimico-fisiche macroscopiche quali la porosità, la densità, il contenuto in sostanza organica, le proprietà del complesso di scambio, assunzione tanto più valida quanto le dimensioni dei serbatoi tendono ad essere piccole. Il serbatoio del profilo, S0, è ipotizzato essere soggetto ad una deposizione meteorica I di inquinante. Il flusso di inquinante Fn che passa da un serbatoio Sn al sottostante Sn+1 è, assunto essere direttamente proporzionale alla concentrazione dell’inquinante nel serbatoio Sn:
Fn = Kn · Sn-1
La migrazione dell’inquinante attraverso il profilo del suolo è così descritta da una serie di n equazioni differenziali:
1) dS0/dt = I - K0·S0
2) dS1/dt = K0·S0 - K1·S1
3) dS2/dt = K1·S1 - K2·S2
···
n) dSn/dt = S(n-1)·K(n-1) - Kn·Sn
Il sistema di equazioni differenziali che descrive l’evoluzione dell’inquinante nel tempo attraverso il profilo secondo il modello a serbatoi e flussi è un sistema di equazioni differenziali lineari omogenee risolvibile. La soluzione generale del sistema può essere espressa nella forma ricorsiva:
S(1,t) = I/K(1) - (I/K(1) - S(1,0))exp (-K(1) t)
S(n,t) = I/K(n) + [(m=1m=n) (K(n-1)/K(n)-K(m))·A(m,n-1)exp(-K(m)·t)] -
- [I/K(n) - S(n,0) + (m=1m=n) (K(n-1)/(K(n)-K(m))·A(m,n-1)]exp(-K(n)t)
dove:
A(1,1) = S(1,0) - I/K(1)
A(m,n) = [S(n,0) - I/K(n) - (m=1
m=n) (K(n-1)/(K(n)-K(m))·A(m,n-1)]exp(-K(n)·t)
I = immissione antropica nel primo strato di suolo;
S(n,0) = concentrazione dell’inquinante nel serbatoio n al tempo zero;
S(n,t) = concentrazione dell’inquinante nel serbatoio n al tempo t:
Secondo il modello proposto, la lisciviazione dell’inquinante dal serbatoio Sn è descritta da una specifica costante cinetica Kn del primo ordine. La costante Kn può essere determinata con procedimenti numerici, quando è nota la deposizione meteorica I, il contenuto dell’inquinante nel serbatoio n al tempo zero S(n,0) ed il contenuto dell’inquinante nel serbatoio n al tempo t, S(n,t).
La costante Kn in termini rigorosamente propri della modellistica è una costante empirica di un modello matematico, che in quanto tale, simula il fenomeno della lisciviazione per analogia con il fenomeno da descrivere, ma senza essere in relazione con i meccanismi chimico-fisici che lo determinano. Nel limite imposto da alcune approssimazioni, si può dimostrare che essa è strettamente legata ad alcuni particolari fenomeni fisici ed alcune particolari proprietà chimiche del suolo.
La costante Kn, è, per definizione, la frazione di inquinante che abbandona lo strato Sn nell’unità di tempo. Essa pertanto può essere considerata avere lo stesso significato di una costante di una cinetica di decadimento del primo ordine. L’assunzione che l'allontanamento di un inquinante da uno strato di suolo obbedisca ad una cinetica del primo ordine è ragionevole quando la velocità con cui l’inquinante diffonde e si omogeneizza all’interno dello strato è molto maggiore rispetto a quella con cui ne è allontanato. La cinetica con cui l’inquinante esce dal serbatoio Sn è infatti esattamente uguale a quella che descrive l’uscita di una specie chimica da un matraccio mantenuto omogeneo da un agitatore magnetico nel quale si ha in entrata un piccolo flusso F l/s di acqua deionizzata ed un uguale flusso F l/s in uscita di acqua contaminata. Tra i processi pedologici che tendono ad omogeneizzare l’inquinante all’interno di uno strato di suolo si possono ragionevolmente annoverare la diffusione dell’inquinante all’interno della soluzione circolante che riempie i pori del suolo in occasione delle piogge e dei successivo periodo in cui il suolo permane umido, la periodica inversione del flusso d’acqua che attraversa la sezione di suolo quale risultato dell’andamento climatico e stagionale delle temperature, delle precipitazioni ed dell’evapotraspirazione, nonché, infine, la bioturbazione operata da organismi animali e vegetali.
La costante Kn può essere utile per stimare approssimativamente la velocità di lisciviazione media attraverso lo strato Sn di spessore h. Infatti se nello strato Sn è mantenuto chimicamente omogeneo dai processi pedogenetici, se nell’intervallo di tempo dt percola la frazione di inquinante Kn , e tutti gli atomi di inquinante hanno la stessa velocità di lisciviazione media Vn, allora la velocità di traslocazione media attraverso lo strato sarà:
Vn Kn · hn
Dalla costante cinetica Kn dello svuotamento dello strato di suolo Sn per analogia con le cinetiche di decadimento del primo ordine si può facilmente calcolare il tempo di semisvuotamento del serbatoio Sn, una costante analoga al tempo di dimezzamento delle cinetiche chimiche del primo ordine:
t(1/2)n = ln2·Kn-1
t(1/2)n è un parametro empirico efficace nel riassumere la velocità di lisciviazione di un inquinante da uno strato di suolo, in quanto indica il tempo necessario affinché la sua concentrazione si dimezzi.
3.7.3.4.
Tempo di residenza.
Il tempo di residenza Trn di un elemento nel serbatoio Sn è definito come il rapporto tra la quantità di elemento immagazzinata nel serbatoio ed il flusso in uscita Kn*Sn. Il tempo di residenza Trn è pertanto uguale al reciproco della costante Kn.
4.
RISULTATI
Per comprendere la sorte degli elementi in traccia che raggiungono il suolo è necessario considerare l’ambiente chimico in cui si vengono a trovare e le reazioni a cui prendono parte. Queste dipendono ovviamente dalle proprietà chimico-fisiche dei suoli, dai processi pedogenetici in atto e degli effetti che l’attività antropica ha nel modificare il pedoambiente. Vengono illustrate e discusse le proprietà chimico-fisiche dei suoli, riservando particolare attenzione a quelle reazioni e a quei processi cui sono soggetti Pb, Cd, Zn, Cu, Cr, Bi, As ed Sb, e che ne possono determinare l’entità del trasferimento entro la biomassa vegetale e la lisciviazione verso la falda.
Le proprietà chimico-fisiche di un suolo sono condizionate sia dalla storia quaternaria del sedimento su cui il suolo evolve sia dall’uso che del suolo è fatto. Caratteristiche geomorfologiche ed uso del suolo, nell’area di Villadossola, sono strettamente legate. E’ ragionevole pertanto supporre che le proprietà chimico-fisiche del suolo siano differenti nelle diverse unità geomorfologiche distinte nel corso del campionamento e che la lisciviazione, la biodisponibilità, o, genericamente parlando, la vulnerabilità del suolo all’inquinamento da metalli pesanti sia significativamente differente nelle diverse unità.
L’osservazione di una vulnerabilità del suolo all’inquinamento significativamente diversa in rapporto alla geomorfologia ed all’uso del suolo può permettere una più corretta gestione del territorio, minimizzando gli effetti dell’inquinamento e permettendo una migliore tutela della salute pubblica.
Come è noto le proprietà chimico-fisiche del suolo possono essere considerate il risultato dei processi pedogenetici quali determinati dai fattori pedogenetici sintetizzati dall’equazione di Jenny (Violante P. 1986):
Suolo = f
(roccia madre, tempo, topografia, clima, vegetazione, antropizzazione)
. Nella discussione che segue la descrizione dei profili particolare riguardo è dato all’analisi di come le
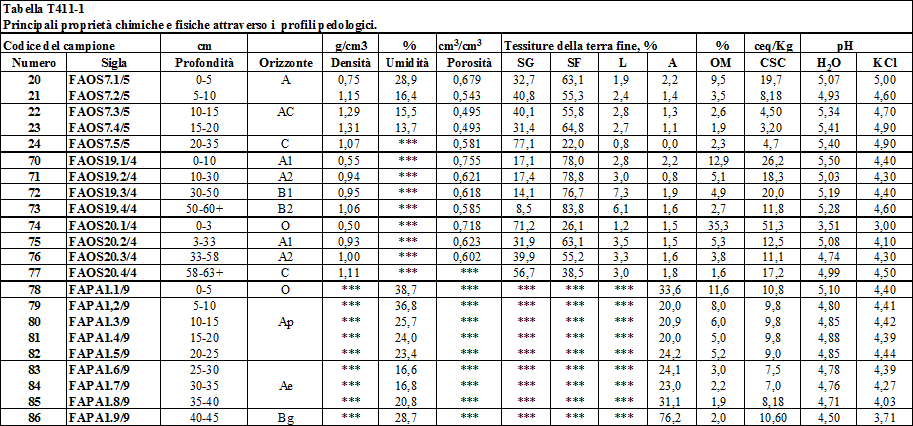
Tabella T411-1. Principali proprietà
chimiche e fisiche attraverso i profili pedologici.
Tabella T411-2. Elementi maggiori totali nei profili pedologici.
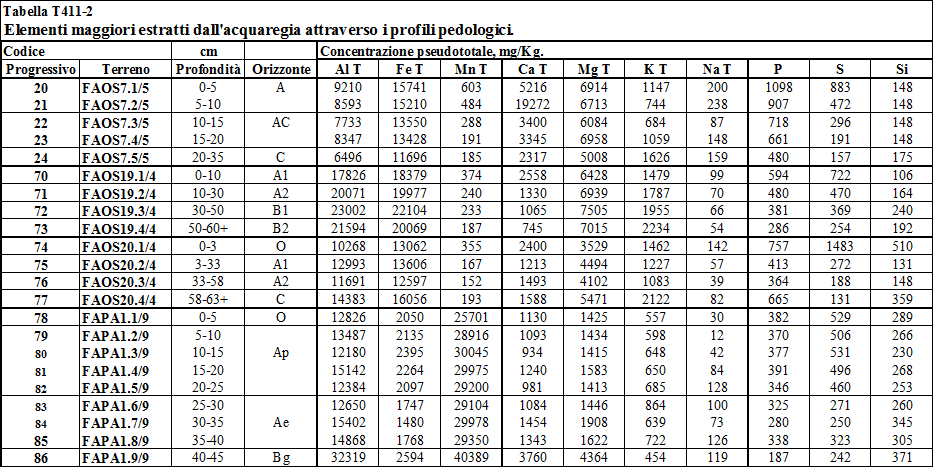
Tabella
T411-3. Elementi in traccia totali nei profili pedologici.
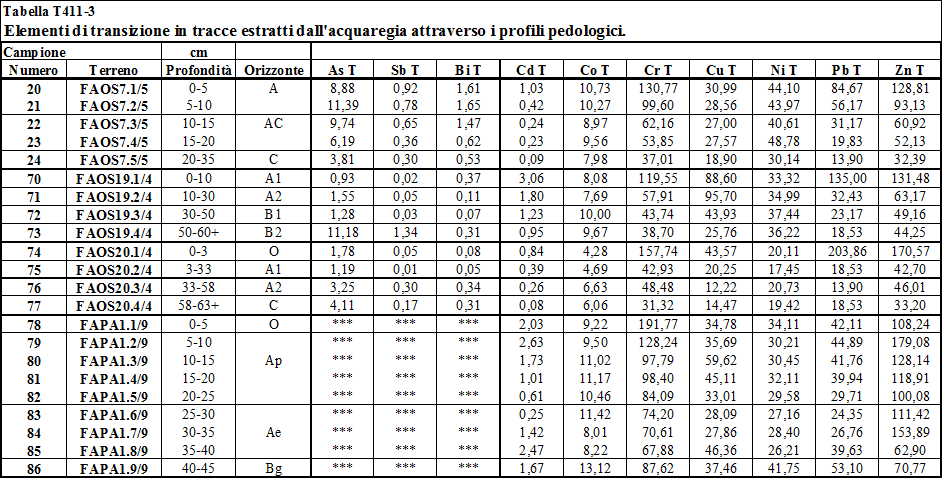
Tabella T411-4. Metalli pesanti estratti dl reagente di Lakanen.
diverse caratteristiche osservate nei profili pedologici siano il risultato della diversa intensità con cui hanno agito i fattori ed i connessi processi pedogenetici. La scelta di questo schema di discussione trova la sua ragione nel presupposto che il metallo inquinante una volta raggiunto il suolo ne entri a far parte del sistema chimico, si misceli all’elemento litogenico e di questo segua il comportamento secondo l’effetto che i fattori e i processi in atto determinano. Le principali proprietà chimiche e fisiche dei profili pedologici sono riportate nelle tabelle T411-1, T411-2, T411-3 e T441-4.
Profilo FAPA1.
Il profilo è stato raccolto nella contea di Cliland, Glasgow Scozia, sulla sommità pianeggiante di un Drumlins alto una trentina di metri e lungo diversi hm. La roccia madre su cui si sviluppa il suolo è un deposito glaciale lasciato all’inizio dell’Olocene (10000 anni fa) dal ritiro del ghiacciaio flandriano. La giacitura immerge di circa il 5% in direzione S-S-W. La carta pedologica del Regno unito lo classifica come Glaysol.
Orizzonte O, 0-5 cm. Colore umido bruno scuro, associato dall’accumulo di spoglie organiche. La densità dell’orizzonte è molto bassa. Le radici delle foraggiere formano un feltro pressoché continuo, la struttura del suolo è poliedrica irregolare, con aggregati sub-centimetrici cementati dalla sostanza organica e dalle radici delle foraggiere. L’orizzonte O deve la sua formazione all’impostazione del prato permanente avvenuta nel 1984, quando il prato permanente è succeduto all’orzo. Il contenuto di argilla dell’orizzonte è sensibilmente superiore a quello del sottostante, suggerendo un tasso di formazione delle argille del 1,3% all’anno. Il limite con l’orizzonte inferiore è lineare e sfumato.
Orizzonte Ap, 5-25 cm. Il colore del suolo umido è bruno-giallo. Il contenuto di sostanza organica dell’orizzonte Ap è inferiore a quello dell’orizzonte organico e decresce esponenzialmente con la profondità. Alla diminuzione del contenuto di sostanza organica si accompagna la diminuzione dell’attività biologica, rappresentata dalle radici delle foraggiere e dagli anellidi. La struttura del suolo è poliedrica irregolare, con aggregati cementati dalla sostanza organica e dalle radici che raggiungono dimensioni centimetriche. Lo scheletro presente nell’orizzonte è costituito da granuli monominerali di quarzo fortemente arrotondati e segnati dalla corrosione chimica. A causa delle lavorazioni agricole la distribuzione dello scheletro attraverso l’orizzonte è fortemente disomogenea. Il limite con l’inferiore sottostante è netto e lineare, segnato dalla soletta di aratura.
Orizzonte Ae, 25-40 cm. Il colore del suolo umido è giallo-bruno, indice della precipitazione di idrossidi di Fe. Il contenuto di sostanza organica e l’attività biologica sono modeste, e decrescono attraverso l’orizzonte. La struttura è poliedrica regolare, gli aggregati sono rivestiti di “cutens”, pellicole di minerali argillosi isoorientati, indice della traslocazione delle argille dall’orizzonte soprastante. Il contenuto di argilla dell’orizzonte Ae è inferiore a quello degli orizzonti sopra- e sottostanti ed aumenta esponenzialmente con la profondità.
Orizzonte Bt, 40-45+ cm. Il
colore del suolo umido è rosso scuro, attraversato da screziature verdastre,
indici di precipitazione di ossidi di ferro e condizioni riducenti. Il contenuto
di argille è molto elevato. La struttura è poliedrica regolare prismatica. Gli
aggregati prismatici sono ricoperti da pellicole isoorientate di argille,,
indice della illuviazione di questa frazione tessiturale dall’orizzonte
soprastante. L’orizzonte ha un’elevatissima umidità relativa. L’attività
biologica è bassissima, rappresentata da rarissimi di dimensioni millimetriche.
Profilo FAOS19.
Il profilo è ubicato su una spalla glaciale del versante destro della valle, con pendenza del 10% circa. L’esposizione è a ESE. Il suolo è sviluppato sul deposito colluviale impiegato nel terrazzamento agricolo del versante, ben preservato ed in uso. In passato l’appezzamento, come alcuni campi limitrofi, per l’idoneo microclima testimoniato da specie xerofile (fico d’india) in stato di deficit idrico, ha ospitato la coltura della vite, ma al momento del campionamento è destinato a prato permanente sfalciato.
Orizzonte A1, 0-10 cm. Colore del suolo umido 10YR 3/3 (marrone scuro). Lo scheletro è scarso e la tessitura sabbiosa (95%), con prevalenza di quella fine (78%). La struttura è poliedrica irregolare, a scala millimetrica, moderatamente resistente. I singoli poliedri sono riuniti in fragili aggregati centimetrici dalle radici delle foraggiere pratensi. Il suolo è poco addensato, molto poroso e di bassa densità. L’attività biologica è elevata e sono abbondantemente presenti le radici fini e i peli radicali. Il limite con l’orizzonte sottostante è diffuso e ondulato.
Orizzonte A2, 10-30 cm. Colore umido 10YR 3/4 (giallastro scuro). Lo scheletro è comune e grossolano, la tessitura è sabbiosa (95%). La struttura è poliedrica sub-angolare, con aggregati che talvolta raggiungono dimensioni di 2-3 cm, moderatamente consistenti. L’attività biologica è media e sono comuni le radici delle foraggiere, che raggiungono i 2 mm di diametro. La porosità e la densità è sono medie. Il limite con l’orizzonte sottostante è graduale e ondulato.
Orizzonte B1, 30-50 cm. Colore del suolo umido 10YR 3/4 (giallastro scuro). La frazione scheletrica è poco rappresentata e prevalentemente composta dagli elementi fini. La tessitura è sabbiosa (91%). La struttura è simile a quella dell’orizzonte sovrastante, poliedrica sub-angolare con aggregati di dimensioni di 2-3 cm di diametro, moderatamente consistenti. L’attività biologica è simile a quella dell’orizzonte soprastante, dove sono comuni le radici di diametro millimetrico. La porosità e la densità sono medie. Il limite con l’orizzonte inferiore è diffuso e discontinuo.
Orizzonte B2, 50-60+ cm. Il colore del suolo umido è 5YR 4/4 (rossastro marrone). Come nell’orizzonte B1 lo scheletro è poco rappresentato ed è principalmente costituito dai termini più fini. Simile è inoltre la tessitura, la densità e la porosità. Sensibilmente inferiore è l’attività biologica in quanto sono visibili solo poche radici.
Profilo FAOS20.
La stazione di campionamento è ubicata sul versante sinistro della valle, su una spalla glaciale. La giacitura dell’appezzamento è sub-pianeggiante e l’esposizione è WNW. L’appezzamento è relativamente poco soleggiato e fresco. L’appezzamento campionato faceva parte di un terrazzamento destinato alla coltivazione della vite. Attualmente il campo è in stato di abbandono ed è in corso un processo di riforestazione spontanea che ha condotto alla formazione di un giovane bosco misto, favorito da un idoneo microclima umido, a prevalente pino, castagno e larice. Sotto la copertura arborea che intercetta una buona frazione dei raggi solari cresce l’erica, alla quale si deve l’accumulo di una spessa lettiera.
Orizzonte O, 0-5 cm. Orizzonte organico costituito nei primi millimetri di spessore da spoglie organiche indecomposte, principalmente foglie di erica e di castagno, e nello spessore sottostante da residui organici progressivamente decomposti. Lo scheletro è assai poco rappresentato e la tessitura è sabbiosa (93%) con prevalenza della frazione grossolana (71%). La sostanza organica ammonta al 35% e l'orizzonte ha una densità molto bassa. L’attività biologica è principalmente determinate dalle ife fungine in quanto le radici sono assenti. I limite con l’orizzonte sottostante è netto e lineare.
Orizzonte A1, 5-35 cm. Colore del suolo umido 7.5YR 3/4 (marrone scuro). E’ abbondante lo scheletro minuto, la tessitura è sabbiosa e principalmente composta dalla frazione fine. La struttura è granulare, con granuli che raggiungono dimensioni di 5 cm, fragili, cementati dalle ife fungine. Il suolo è poroso e poco addensato. La sostanza organica è abbondante e in parte costituita da spoglie organiche indecomposte. L’attività biologica è elevata, e accanto alle radici ed ai peli radicali si osservano ife fungine. Il limite con lo strato sottostante è graduale ed ondulato.
Orizzonte A2, 35-60 cm. Colore del suolo umido 5YR 3/2 (marrone rossastro scuro). Abbondante scheletro da fine a medio, tessitura sabbiosa con prevalenza di quella fine. Struttura granulare irregolare, a grana singola, con aggregati di dimensioni millimetriche. Porosità e densità medie. Modesta attività biologica, determinata prevalentemente da scarse radici millimetriche. Non sono visibili ife fungine. Il limite con l’orizzonte sottostante è diffuso e ondulato.
Orizzonte C, 55-60+ cm. Colore del suolo umido 7.5YR 4/6 (marrone intenso). Scheletro molto abbondante, con clasti a spigoli vivi che raggiungono un diametro di alcuni cm. Tessitura sabbiosa, con prevalenza della frazione grossolana (56%). Non sono evidenti aggregati, il suolo è sciolto ed i singoli granuli minerali sono ben separati. Il contenuto di sostanza organica è basso e la densità è elevata. Attività biologica molto bassa, radici assenti.
Il suolo è ubicato sulle alluvioni di fondovalle e ha quindi giacitura pianeggiante. Il sedimento su cui si sviluppa il suolo è costituito da lenti di sabbie fini e ciottoli deposte dalle alluvioni del fiume Toce. L’appezzamento è destinato all’avvicendamento del prato con le colture cerealicole.
Orizzonte A, 0-10 cm. Il colore del suolo umido è 10YR 3/2, (grigiastro marrone molto scuro).Il suolo è privo di scheletro e molto sabbioso (91% di sabbia) e privo di plasticità. La porosità è elevata e, a eccezione di aggregati cementati dalle radici fini, il suolo è privo di struttura. L’attività biologica è molto elevata, e si osservano molto abbondanti le radici e i peli radicali delle foraggiere pratensi. Il limite con l’orizzonte sottostante è netto.
Orizzonte AC, 10-20 cm. Il colore del suolo umido è 10YR 5/2, (grigiastro marrone) con screziature 7.5YR 4/6 (marrone intenso). Lo scheletro è poco abbondante. La tessitura è più ricca in sabbia dell’orizzonte A (96% contro 91%) e più povera in frazione fine (3,8 contro il 3,2%). Le sabbie, il principale costituente tessiturale, sono più addensate che nell’orizzonte superficiale. Sono evidenti aggregati poliedrici irregolari a grana singola molto deboli. L’attività biologica è modesta e si osservano poco abbondanti radici fini. Il limite inferiore dello strato è netto, e corrisponde alla suola di lavorazione.
Orizzonte C, 20-35+ cm. Il colore è 2,5Y 5/2 (grigiastro marrone). Lo scheletro è abbondantemente rappresentato. Esso è costituito da ciottoli eterodiametrici addensati con diametro superiore ai 3 cm. La sabbia grossa è la principale componente tessiturale della frazione fine. Limo ed argilla sono pressoché assenti. L’attività biologica è molto scarsa, ed è principalmente costituita da radici di qualche millimetro di diametro.
La composizione chimica e mineralogica della roccia madre su cui si sviluppano i suoli è in prima approssimazione simile per tutti i suoli campionati, essendo essa costituita da un deposito morenico di età olocenica, prodotto dall’esarazione di settori crostali ampi diverse decine (Villadossola) o centinaia (Cliland) di Km (Villadossola). Il fattore tempo è anch’esso approssimativamente lo stesso per la Scozia e l’Italia, essendo in entrambe i casi la roccia madre costituita da una morena deposta 10000 anni fa. Poco è noto sulla vegetazione che si è sviluppata sui suoli considerati. E’ ragionevole ritenere che con il procedere della deglaciazione determinata dalla precessione dell’asse di rotazione terrestre si siano succedute, con un certo sfasamento determinato dalla latitudine, biocenosi tipiche del deserto artico, della taiga, della tundra, foreste di conifere ed infine di latifoglie. In tempi storici più o meno recenti le foreste infine sono state tagliate ed i suoli, antropizzati, destinati all’agricoltura e all’uso residenziale. Ricostruire con maggior dettaglio l’evoluzione della vegetazione sui suoli considerati e se questa abbia determinato le proprietà del suolo che sono oggi osservate, per quanto affascinante, esula dagli scopi della presente indagine. Quanto si osserva dell'attività biologica è l’uso recente del suolo: prati in rotazione agricola con colture cerealicole (FAOS21), prati permanenti sfalciati (FAOS19, FAPA1) e suoli sviluppati sotto bosco (FAOS20). La vegetazione che si sviluppa sul suolo ha un ruolo notevole nel determinare l’accumulo di sostanza organica osservato. Il suolo scozzese ed il suolo italiano si differenziano nettamente per il fattore tettonico. Diverso è l’ambiente geotettonico su cui si sviluppa il suolo scozzese ed il suoli italiano. Il profilo di Cliland è ubicato su un settore crostale soggetto ad un lento sollevamento eustatico, stimato in ragione di 1-4 mm/a (Cameron B.I. e Stephenson D., 1985), mentre i suoli di Villadossola sono ubicati sull’arco alpino, dove la velocità di sollevamento e di erosine sono stimate nell’ordine di grandezza del cm/a. La differente velocità di erosione è un fattore che contribuisce a spiegare la grande differenza nel contenuto di argille del suolo scozzese e dei suoli di Villadossola. I suoli di Villadossola sono ricchi in scheletro e sabbia costituite da frammenti litici o di minerali facilmente alterabili come serpentino e feldspati, e sono poveri in limo e argille ad indicare un limitato sviluppo pedogenetico. Il basso contenuto di argille dei suoli di Villadossola è la conseguenza dall’elevata energia di scorrimento delle acque superficiali. Tuttavia l’erosione selettiva delle argille e del limo spiega le differenze tessiturali tra i suoli di Villadossola ma non è un processo che da solo possa spiegare le differenze tessiturali, mineralogiche e strutturali tra il suolo scozzese ed i suolo alpino, in parte attribuibile alle differenti condizioni climatiche. La combinazione delle temperature e delle precipitazioni fa sì che i suoli delle Alpi siano soggetti a brevi periodi di intense precipitazioni in grado di causare eventi alluvionali, lunghi periodi di deficit idrico e di gelo, periodi in cui i processi di formazione del suolo sono rallentati dal modesto contenuto d’acqua del suolo. Lo scarso sviluppo dei suoli di Villadossola rispetto a quello di Cliland va quindi attribuito sia ad una maggiore intensità con cui insiste l’erosione, che al regime climatico, poco favorevole alla formazione ed alla conservazione in situ dei minerali argillosi pedogenetici.
I principali dati analitici dei suoli di Villadossola e le caratteristiche della stazione di campionamento sono riportati in appendice (A1-A7). Nelle tabelle T413-1, T413-2, T413-3 e T413-4 sono invece illustrate le medie e i coefficienti di variazione standard delle proprietà chimico-fisiche dei profili distinti per profondità dello strato e quindi anche per uso, stazione geomorfologica e pratiche di gestione attuali. Le tabelle permettono in prima approssimazione di determinare se tra le classi considerate sussistono differenze statisticamente significative tenendo conto del numero dei campioni presenti nella classe, della media e della deviazione standard. In prima approssimazione si può ritenere che tra due classi vi siano differenze significative con un limite di confidenza superiore al 60% quando la differenza tra le due medie è superiore alla deviazione di almeno una delle due classi.
La tessitura del suolo è una caratteristica importante nel determinare proprietà del suolo che possono spiegare la mobilità dei metalli pesanti provenienti dalle emissioni fusorie della fonderia. Tra queste vi sono principalmente la permeabilità e la capacità di scambio cationico. Queste caratteristiche fisiche e chimiche del suolo hanno grande rilievo nel determinare la velocità di lisciviazione degli inquinanti verso la falda e la loro disponibilità nei confronti degli organismi vegetali e animali.
La tessitura è prevalentemente sabbiosa in tutti i suoli considerati. In media si
misura il 25% di sabbia grossa, il 66% di sabbia fine, il 6% di limo ed il 3% di argilla.
La tessitura dei suoli di Villadossola è il risultato di fattori pedogenetici, la cui azione può essere importante nel determinare il comportamento e la mobilità degli elementi in traccia. Tra questi i più importanti sono la neoformazione di argille e la loro traslocazione verso gli strati più profondi del suolo; la formazione di aggregati di suolo ad opera di sostanze cementanti quale la sostanza organica; il rimodellamento superficiale ad opera dello scorrimento delle acque superficiali, che ridistribuisce il materiale terrigeno eroso dalle porzioni più acclivi secondo la pendenza locale.
In
nessuna delle unità pedoambientali considerate si osserva una significativa
differenza tessiturale attraverso il profilo del suolo. In particolare non si
osserva nessun significativo arricchimento della frazione argillosa negli
orizzonti profondi neanche nel profilo dei suoli indisturbati, dove i processi
pedogenetici che portano alla neoformazione di argilla e alla sua traslocazione
negli orizzonti profondi hanno agito per i tempi più lunghi e sono pertanto più
pronunciati. Non si osserva inoltre nessuna differenza tessiturale
significativa tra gli orizzonti delle diverse unità d’uso del suolo, dove le
differenze nel contenuto in sostanza organica sono massime. Queste osservazioni
permettono di concludere che in nessuna delle unità pedoambientali considerate
vi è un apprezzabile fenomeno di illuviazione delle argille e si può escludere
che la traslocazione dell’inquinante in forme adsorbite sui minerali
dell'argilla sia un fenomeno di rilievo nel determinare la distribuzione
verticale degli inquinanti. Si può inoltre escludere che l’azione cementante
della sostanza organica affligga la determinazione della tessitura con erosi
sistematici significativi.
Tabella T413-1.
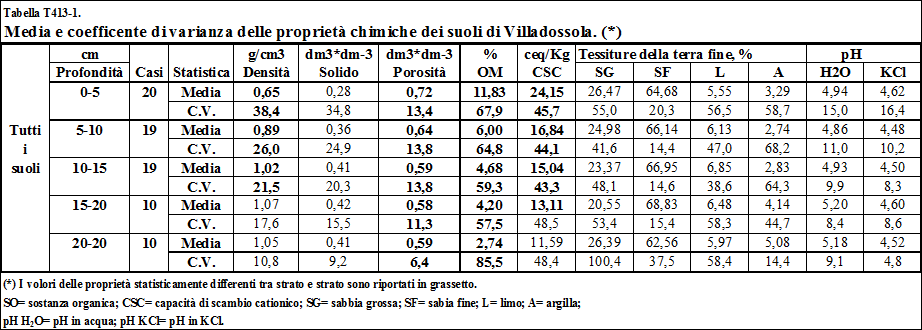
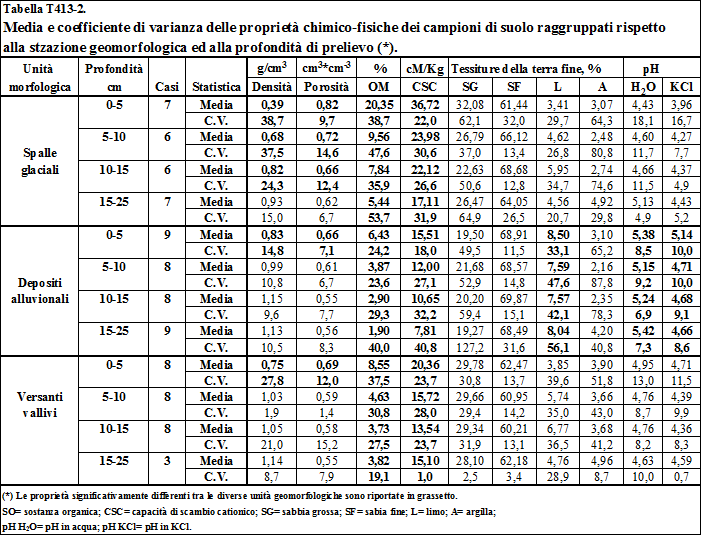
Tabella T413-2.

Tabella T413-3.

Tabella T413-4.
Dall’idrogeologia è noto che la permeabilità dei suoli controlla sia la frazione di acque piovane che si infiltra nel sottosuolo (infiltrazione efficace) che la velocità con cui questi fluiscono attraverso il profilo (Francani V., 1988. Frega G., 1987. Maione U., 1995. Terzaghi P., 1967. Feddes R. A. et al., 1988). Infiltrazione efficace e permeabilità dipendono dalla tessitura e dalla struttura del suolo. In prima approssimazione la struttura del suolo può essere trascurata. Infatti si osserva che la permeabilità e l’infiltrazione efficace aumentano progressivamente con il contenuto in frazione grossolana. Oltre alla composizione tessiturale del suolo è quindi importante la distribuzione geometrica e i rapporti tra le particelle minerali del suolo, ovvero la sua struttura. Delle diverse classi tessiturali presenti nei suoli di Villadossola è ragionevole ritenere che la permeabilità e quindi l’infiltrazione efficace del suolo siano controllate principalmente dalla sabbia fine e dal limo. Lo scheletro e la sabbia grossa sono infatti rappresentate nei suoli in quantità troppo esigue per poterne determinare la permeabilità, ed è atteso che queste classi tessiturali si comportino nei confronti della soluzione circolante come una massa inerte immersa in una matrice più fine. Il contenuto di argilla è per contro eccessivamente esiguo per riempire completamente le cavità e gli interstizi lasciati liberi dall’impacchettamento dei granuli di sabbia fine e questa frazione tessiturale non può quindi controllare la permeabilità dei suoli.
A causa dell’elevata attività superficiale e l’adsorbimento di cationi, la frazione fine, limo e argilla, è notoriamente importante anche nel determinare la biodisponibilità dell’inquinante (Adriano D.C., 1986. Alloway B.J., 1997. Sillampää M., 1982). Le argille del suolo possono rimuovere il metallo presente in soluzione adsorbendolo sulle proprie superfici o fissandolo all’interno dei propri interstrati (Bolth G.H., 1963. Bolth G.H., 1955. Dolcater D.L., 1972. Egozy Y., 1980. Lim C.H., et al., 1980. Evans L. J., 1989. Schindler P.W., et al, 1976. Bittel J.F. e Miller R.J., 1974. Farrah H. e Pickering W.C., 1976) diminuendo nella soluzione circolante la concentrazione della forma che più facilmente passa la membrana cellulare dei peli radicali, quella solubile. Inoltre Vermiculiti e Smectiti possono adsorbire all’interno del proprio interstrato i metalli in una forma che non viene rilasciata a contatto con i succhi gastrici dei mammiferi, diminuendone, lievemente, l’assimilazione da parte delle specie animali quale risultato dall’ingestione di suolo (Sheppard S. C. et al., 1994).
Il contenuto in sostanza organica nei suoli di Villadossola è compreso tra 0.7 e 35.3%. In tutte le unità pedologiche considerate il contenuto di sostanza organica è massima negli strati superficiali e diminuisce a valori minimi in quelli più profondi (tabelle T413-1, 2, 3 e 4). Il profilo della sostanza organica è ovviamente controllato dall’uso del suolo come si può apprezzare in figura F4131-1. I
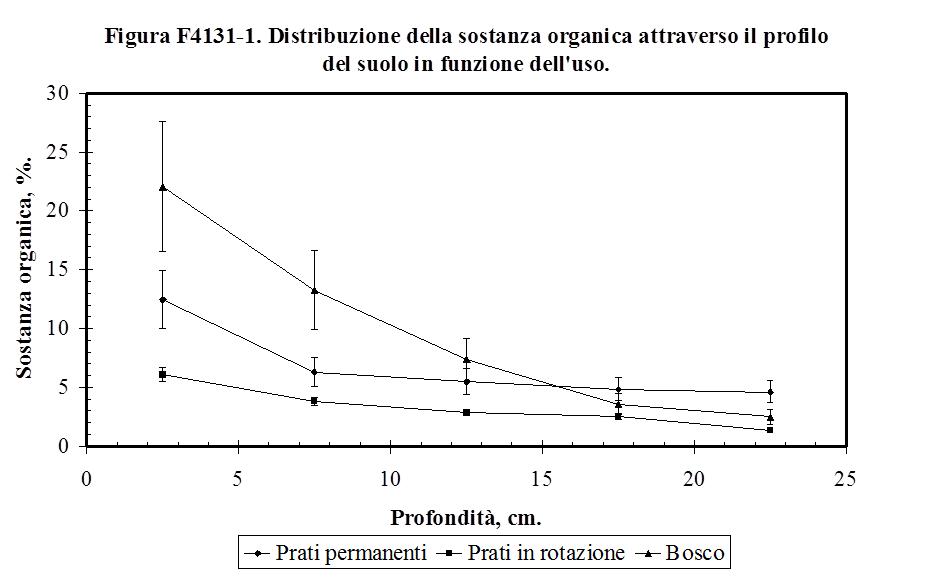
Figura F4131-1. Distribuzione della sostanza organica nel profilo secondo l'uso
del suolo.
contenuti minimi in sostanza organica si osservano nel profilo dei prati lavorati nel corso degli ultimi dieci anni, i valori intermedi si hanno nel profilo dei prati permanenti ed infine i valori massimi si registrano nei suoli forestali. A profondità superiori a quindici centimetri il contenuto di sostanza organica è mediamente compreso tra il 2 ed il 5% e le influenze determinate dall’uso del suolo sono minime. La velocità di degradazione della sostanza organica apportata ai suoli alpini simili a quelli di Villadossola è del 3% annuo (Arduino E. et al., 1986). Considerati i tempi di rinnovo dei prati permanenti e dei boschi e la diffusione che le pratiche agricole di Villadossola hanno in vaste aree dell’arco alpino occidentale, è ragionevole ritenere che, quantomeno nei boschi e nei prati permanenti, il contenuto in sostanza organica osservato sia quello determinato dal raggiunto equilibrio tra apporti asporti e degradazione, abbia raggiunto lo stato stazionario e sia rappresentativo di un’ampia area geografica.
Molti lavori pubblicati da numerosissimi autori indicano che la sostanza organica è un componente del suolo molto importante nel determinare il comportamento dei metalli pesanti nel suolo (Adriano D.C., 1986. Alloway B.J., 1997. Sillampää M., 1982). In particolare è importante per determinarne le forme chimiche, la concentrazione nella soluzione circolante, la velocità di lisciviazione e la biodisponibilità.
La sostanza organica presente nel suolo può forme assai varie, comprese tra le spoglie organiche indecomposte e gli acidi umici e fulvici (Testini C. e Gessa C., 1986. Schnitzer M., 1991). La sostanza organica può quindi complessare il metallo in forme insolubili e rallentarne la lisciviazione o chelarlo in forme solubili e mobilizzarlo. La sostanza organica può inoltre occludere all’interno delle propria struttura (Schnitzer M., 1991) i metalli pesanti rimuovendoli dalla soluzione circolante (Saar R. A., e Weber J.H., 1980), diminuendone sensibilmente, come nel caso del Cu (Sillampää M., 1982) la frazione assimilata dai vegetali. A causa del carattere acido, l’accumulo di sostanza organica può inoltre abbassare il pH del suolo, favorendo così la solubilizzazione dei metalli pesanti occlusi nel reticoli cristallini di ossidi, idrossidi, carbonati e fosfati che possono precipitare solo a pH elevati. I metalli di transizione formano con i radicali dei composti organici, principalmente con R-COO-, R-NH2, R-COOH, R-SH, complessi assai stabili rispetto a quelli formati dai metalli alcalini ed alcalino-terrosi (Yatsimirskii K. B. and Vasil’Ov V.P., 1960. Schwarzen G. and Sillen G. L., 1958).
La CSC dei suoli studiati è compresa tra il valore minimo di 3.2 cmol/kg e il valore massimo di 51.3 cmol/kg. Considerato che il contenuto di argilla è sempre piuttosto basso e che la distribuzione
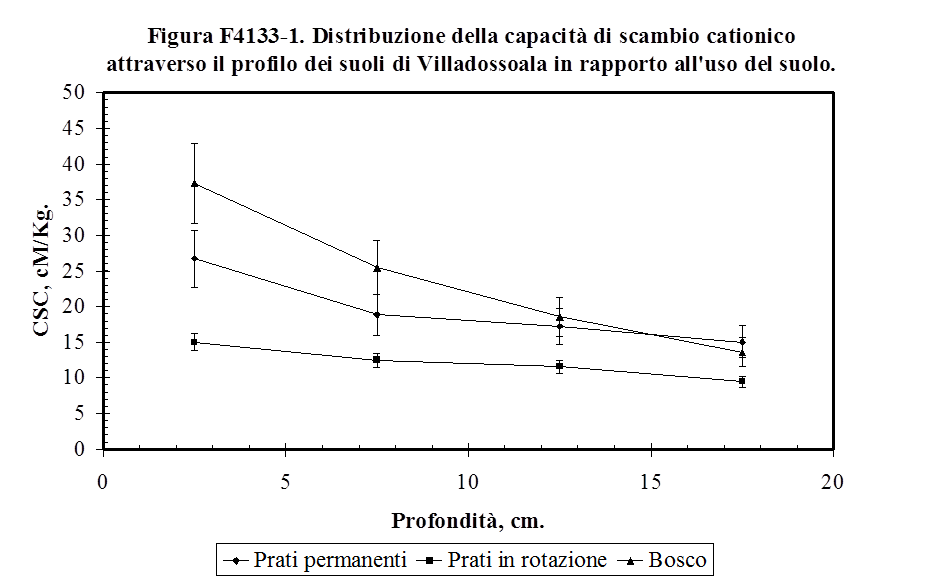
Figura F4133-1. Distribuzione della CSC nel profilo delle unità d'uso del
suolo.
delle frazioni tessiturali è relativamente omogenea sia tra gli strati di uno stesso profilo che tra i profili delle diverse unità pedologiche distinte nel corso del campionamento, è ragionevole ritenere che la distribuzione della CSC sia determinata principalmente dalla sostanza organica. Invero la distribuzione della CSC attraverso il profilo grossomodo riflette quello della sostanza organica (figura F4133-1). Tra gli strati di un profilo e tra i profili delle diverse unità d’uso del suolo si osservano notevoli differenze nei valori di CSC (T413-4). I valori minimi di CSC si osservano così nei suoli a prato avvicendati con le colture cerealicole, i valori intermedi nei profili dei prati permanenti ed i valori massimi nei profili dei suoli forestali. La CSC aumenta dove le pratiche agricole conducono all’accumulo di sostanza organica ed è quindi una proprietà chimica del suolo fortemente controllata dall’attività antropica. A profondità superiori ai 15 cm, dove diminuisce l’influenza delle pratiche agricole, la CSC converge a valori più bassi.
A determinare la CSC del suolo oltre che i colloidi organici concorrono ovviamente anche le superfici minerali. Le superfici dei minerali del suolo sono infatti elettricamente cariche e chimicamente reattive (Gessa G., Testini C., 1989b). Smectiti, Vermiculiti, Idromiche, Illiti e ossidi e idrossidi di Fe e Mn possono adsorbire i metalli, rimuovendoli dalla soluzione circolante rallentandone la lisciviazione verso gli strati profondi del suolo o limitandone la biodisponibilità. Siccome la superficie dei minerali del suolo aumenta in modo inversamente proporzionale al quadrato del raggio medio della particella, le frazioni più reattive nel complessare i metalli sono quella argillosa e limosa. Le superfici minerali e i composti umici hanno inoltre una carica pH-dipendente affatto diversa ed inoltre si combinano con gli elementi in traccia formando complessi dotati di una stabilità molto diversa. Il complesso di scambio determinato dalla sostanza organica e quello determinato dalle superfici dei minerali del suolo ha quindi un comportamento chimico nei confronti dei metalli pesanti molto differente.
La CSC risulta quindi fortemente correlata sia alla sostanza organica (SO) che alla frazione fine, limo + argilla (F4133-2):
CSC = 1.49 SO + 0.244 F + 5.07 (p <0.001; r2 =
0.87; n=77)
La frazione fine e la sostanza organica, come è evidente dal coefficiente di determinazione, spiegano l’87% della varianza della CSC. Si può presumere che il rimanente possa essere spiegato dalla composizione mineralogica della frazione fine in particolare dal contenuto di Vermiculiti (cfr. paragrafo Mineralogia dei suoli di Villadossola).
Figura
4133-2. Rapporto tra CSC e sostanza
organica.
Tabella T4133-1. Capacità di scambio cationico della sostanza organica e della frazione
minerale nelle diverse unità geomorfologiche e d'uso.

Tabella T4133-1. Capacità di scambio cationico della
sostanza organica e della frazione minerale nelle diverse unità geomorfologiche
e d'uso (continua).

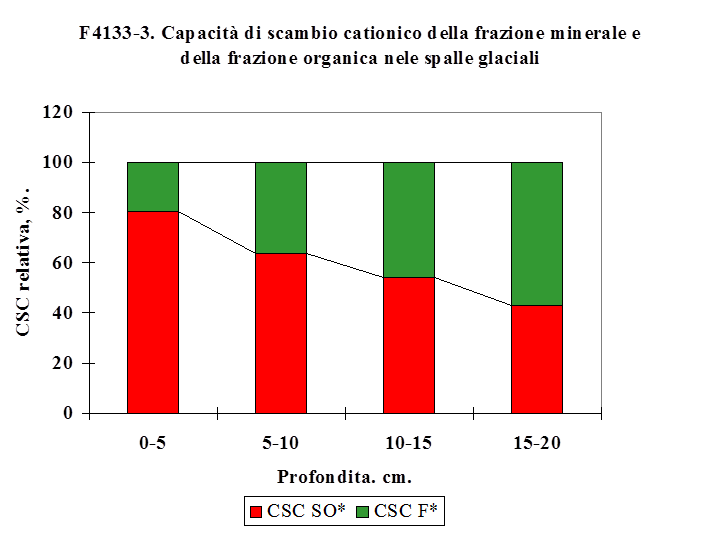
Figura F4133-3. Capaità di sambio
cationico relativa della frazione minerale e della sostanza organica nelle
spalle glaciali.
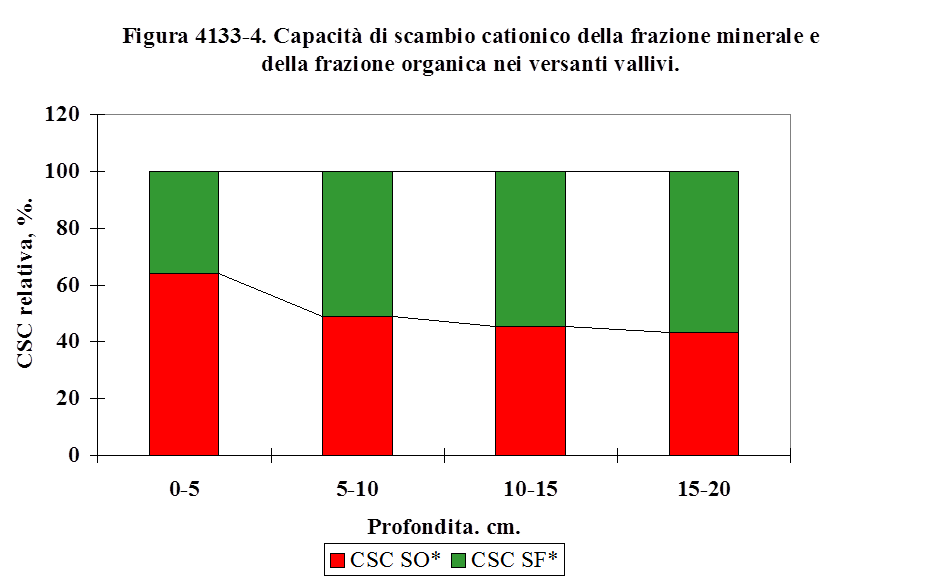
Figura F4133-4. Capaità di sambio cationico relativa della frazione minerale e della
sostanza organica nei versanti vallivi.
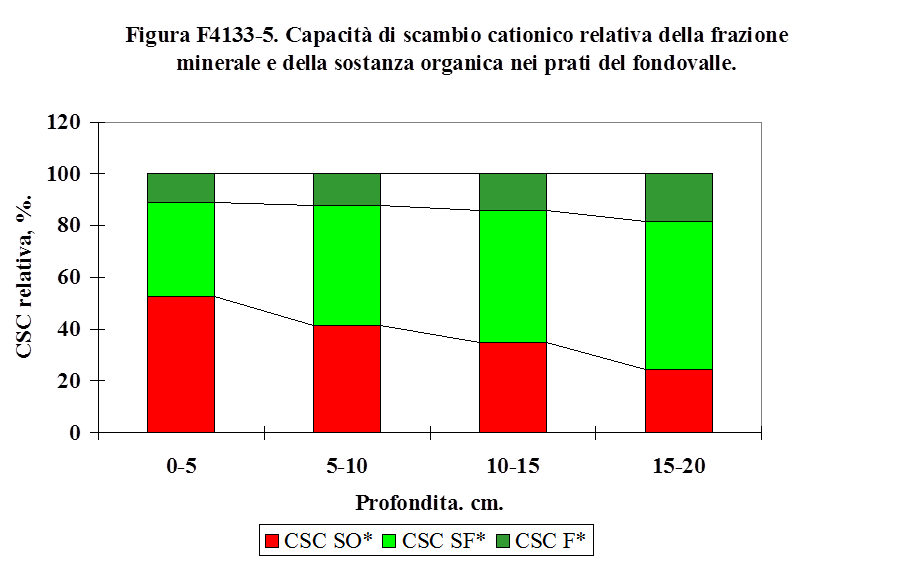
Le capacità di scambio cationico della frazione minerale e della frazione organica sono state calcolate mediante regressioni multiple nelle diverse unità geomorfologiche e d’uso (T4133-1). Differenze significative della capacità di scambio cationico della sostanza organica si osservano in funzione dell’uso del suolo. La capacità di scambio cationico della sostanza organica dei prati permanenti è di 173+/-7,0% cM/Kg, significativamente maggiore a quella dei boschi e dei prati in rotazione agricola, 132+/-16 cM/Kg. Differenze significative nella capacità di scambio cationico della frazione minerale si osservano in funzione dell’unità geomorfologica. Nelle spalle glaciali la capacità di scambio cationico è principalmente determinata dalla frazione limo+argilla ed è di 98+/-40 cM/Kg. Negli acclivi versanti vallivi la capacità di scambio cationico della frazione minerale è principalmente determinata dalla sabbia fine, , ed è di 21+/-23 cM/Kg. Nelle alluvioni del fondovalle la capacità di scambio cationico della frazione minerale è significativamente determinata sia dalla frazione fine limo+argilla (20+/-50 cM/Kg) che dalla sabbia fine (12+/-16 cM/Kg). Le differenze statisticamente significative osservate nella capacità di scambio cationico in funzione della unità geomorfologica permettono, nell’alea del metodo di stima adottato, di tracciare il profilo dell’importanza relativa della sostanza organica e della frazione minerale nelle spalle glaciali, sui versanti vallivi, e nel fondovalle alluvionale (F4133-3, F4133-4, F4133-5). L’importanza relativa della sostanza organica dipende ovviamente dall’uso del suolo prevalentemente associato all’unità geomorfologica: prato in rotazione agricola sul fondovalle, prato permanente o bosco sui versanti vallivi, prato permanente sulle spalle glaciali.
Le differenze osservate nella capacità di scambio cationico della frazione minerale in funzione dell’unità geomorfologica possono essere spiegate dalla velocità di scorrimento delle acque superficiali (Castiglioni G.B., 1986. Birkeland P.W., 1983), che, ad incominciare dall’Olocene, 10.000 anni fa, rimodellano le superfici topografiche di Villadossola. In corrispondenza delle spalle glaciali la velocità di scorrimento delle acque di ruscellamento è bassa e possono pertanto essere deposte solo le particelle fini caratterizzate da una densità bassa, quali le Vermiculiti, le Idromiche e le Illiti. Attraverso gli acclivi versanti vallivi le velocità di scorrimento delle acque superficiali è elevata, e le particelle fini possono essere deposte solo quando hanno una densità elevata come quella del Quarzo, dei Feldspati e dei Pirosseni, minerali aventi una bassa capacità di scambio cationico. Le Illiti, poco dense, possono essere deposte solo quando hanno una diametro superiore a quello del limo. Quando i cristalli di Illite hanno queste dimensioni permangono a lungo nel pedoambiente, potendosi così alterare profondamente ed aumentare la propria capacità di scambio cationico (Ajmone Marsan F. et al., 1988. Arduino E., et al. 1986). Al procedere dell’alterazione della Illite, aumentano sia la capacità di scambio cationico che la selettività verso i cationi a bassa energia di idratazione quali Rb, Cs, K, Pb e Cd. Nel fondovalle la
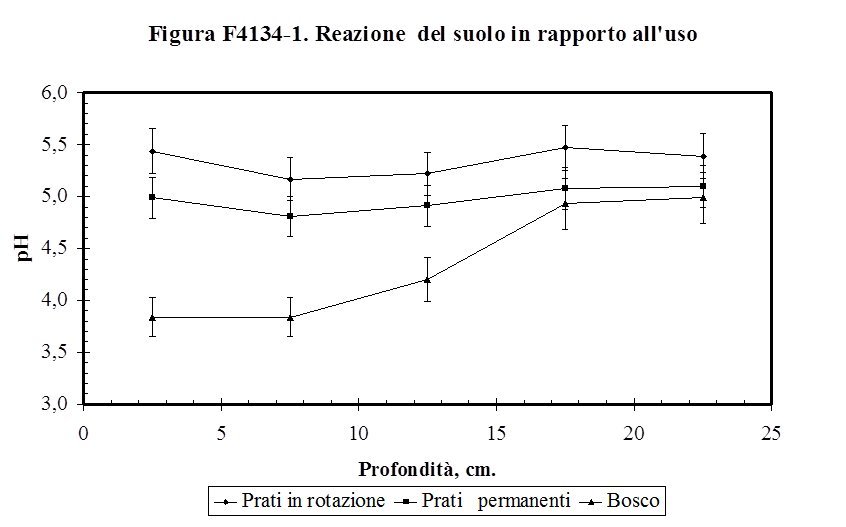
velocità di scorrimento delle acque diminuisce e tornano ad essere deposte le particelle fini dotate di elevata capacità di scambio cationico e le leggere Idromiche sabbiose formatesi sui versanti vallivi dall’alterazione delle miche.
4.1.3.4. Il pH
Il pH dei suoli di Villadossola è compreso tra un valore minimo di 3.5 e il valore massimo di 6.0. L’attività dello ione idronio nella soluzione circolante diminuisce con la profondità in tutte le unità pedologiche considerate (tabella T413-1, T413-2, T413-3, T413-4), sebbene non a profondità superiori ai 15 cm. Vi è una stretta relazione tra uso del suolo, contenuto in sostanza organica e pH (figura F4134-1). L’attività delo ione idronio è massima nel profilo dei suoli forestali, intermedia nel profilo dei prati permanenti ed è minima nel profilo dei prati in rotazione agricola.
Il pH del suolo influisce su molte reazioni e processi chimici che controllano la biodisponibilità e la lisciviazione dei metalli in traccia (Adriano D.C., 1986. Alloway B.J., 1997). In particolare il pH ha un ruolo importante nel determinare la solubilità di fasi minerali che possono occludere al loro interno gli elementi in traccia, sulle forme chimiche che gli elementi assumono in soluzione, sulla carica superficiale dei composti minerali ed umici, sull’intensità dell’adsorbimento e della complessazione e quindi sulla costante di ripartizione tra solido e liquido e sulla velocità di lisciviazione.
4.2.5. Analisi alla microsonda.
Le analisi in microsonda sono state eseguite su aggregati di suolo, detti anche organoliti del campione FAOS1/3. La microsonda permette l’analisi morfologica delle superfici sui campioni rivestiti di grafite e l’analisi chimica quantitativa dei campioni inglobati in resina e lucidati. Le analisi chimiche, morfologiche e microtessiturali effettuate con la microsonda permettono di acquisire utili informazioni sulla composizione mineralogica, sulle caratteristiche della superficie dei minerali, nonché di evidenziare i processi di alterazione in atto sui minerali primari ed alcune importanti caratteristiche microtessiturali importanti nello spiegare la velocità di lisciviazione degli inquinanti e la loro biodisponibilità.
Nelle figure F425-1c e F425-1d è riportato l’immagine in elettroni retrodiffusi di un frammento di roccia costituito da quarzo, feldspati ed idrossidi di ferro. Gli idrossidi di ferro risultano nella immagine di un colore bianco brillante rispetto a quarzo e feldspato in quanto il Fe riflette con maggior efficienza gli elettroni di quanto non facciano gli elementi leggeri (Ca, Na, K, Al, Si) che costituiscono il Quarzo e i Feldspati. Gli ossidi di Fe evidenziano al loro interno una struttura fine, a
F425-1.
F425-2.
bande concentriche, spesso visibile anche ad occhio nudo nelle pseudomorfosi di limonite su solfuri. Le pseudomorfosi di limonite su solfuri non sono comuni nel campione esaminato, e l’ossido di ferro riportato è l’unico rinvenuto. Nella figura F425-1a è riportata la fotografia di un fascio di fibre di serpentino appartenenti alla frazione limosa. Le fibre risultano separate da cavità che si insinuano profondamente nel frammento policristallino. L’origine di tali cavità va ragionevolmente attribuita all’alterazione pedogenetica, in quanto la roccia inalterata, formatasi nelle profondità della crosta terrestre, è caratterizzata da una marcata compitazione delle fibre che non si riscontra nel campione esaminato. Le superfici dei feldspati della frazione limosa (F525-1b, F425-2a, F425-2c) sono subrotondeggianti e non presentano gli angoli vivi che comunemente caratterizzano i feldspati fratturati di fresco. La causa dell’arrotondamento delle superfici dei feldspati va attribuita alla alterazione chimica dei minerali che occorre in ambiente pedogenetico. Infatti le superfici dei granuli di feldspato sono attraversate da profonde cavità subrotondeggianti, segnalate in letteratura come frutto di corrosione chimica nonché da fessure allargate dall’alterazione che seguono i sistemi di sfaldatura e si insinuano profondamente all’interno del cristallo (F425-1b). Le superfici dei piani basali delle miche limo evidenziano processi di sfaldatura paralleli al piano basale (F425-2d e F425-2b). Le dimensioni dei frammenti di mica che si distaccano dai frammenti della frazione limosa, indicati con due crocette nella figura F425-2b, sono quelle dell’argilla. L’origine della defoliazione delle miche è stata attribuita in letteratura (Violante P., 1986) all’azione fisica del gelo e del disgelo nonché all’azione abrasiva esercitata dai minerali più resistenti all’abrasione. L’attività della superficie della frazione limosa è infatti troppo modesta perché i processi di alterazione chimica possano essere marcatamente più incisivi dei processi di alterazione fisica. L’alterazione chimica è al contrario il principale meccanismo di alterazione pedogenetica della frazione argillosa, caratterizzata da un’elevata attività superficiale. I piani lungo i quali due foglietti di mica si distaccano assumono una notevole importanza per spiegare in taluni suoli il comportamento degli elementi in traccia. Lungo tali piani la distanza basale si riduce progressivamente raggiungendo l’intervallo compreso tra i 14 e i 10 Å. Tale zona è detta zona a cuneo. Essa ospita i cosiddetti siti di adsorbimento di “bordo da alterazione” nei quali sono adsorbiti selettivamente cationi a basso potenziale ionico quali Pb, Cs, Pb e Cd.
I granuli di quarzo, non riportati in figura, nonostante abbiano una durezza circa uguale a quella dei feldspati (7.0 nella scala di Mohs) sono caratterizzati da spigoli vivi. Le caratteristiche superfici di frattura concoidi sono perfettamente preservate nonostante i processi di alterazione chimica ed i processi abrasivi che devono aver agito nel corso del trasporto del sedimento. La ragione della pressoché perfetta conservazione delle superfici di frattura dei granuli di quarzo va presumibilmente ricercata nella ben nota resistenza all’alterazione chimica di questo minerale e nella relativa modesta distanza di trasporto dalla cava di prestito. I granuli di quarzo risultano infatti fortemente arrotondati solo a seguito di trasporti di diverse centinaia di Km, come nel caso del loess deposto sulla collina torinese o nelle alluvioni di bassa pianura.
I singoli cristalli ed i rari frammenti di roccia della frazione limosa ed argillosa sono aggregati a formare zollette da una sostanza dall’aspetto mucillaginoso, talvolta filamentoso (F425-3c, F425-3a). In cemento mucillaginoso costituisce una massa amorfa percorsa da fratture di disseccamento che ingloba completamente le componenti minerali e le spoglie organiche indecomposte (F425-3d, F425-3b). Nelle sezioni lucidate (F425-3-c), dove è ovviamente indistinguibile dalla resina a base di carbonio che ingloba le zollette, è evidente che la mucillagine penetra all’interno degli aggregati inglobando completamente le particelle minerali e distanziandole le une dalle altre. Le mucillagini osservate risultano assai diverse dalle immagini dei composti umici provenienti da laghi e suoli pubblicate in letteratura (Schnitzer M., 1991). Queste hanno infatti una forma filamentosa e una struttura reticolare. E’ quindi lecito ipotizzare che la sostanza organica che si osserva inglobare la frazione minerale possa essere costituita da mucillagini radicali ed essudati radicali.
Le osservazioni effettuate con la microsonda permettono di trarre importanti risultati. Mentre i frammenti litici sono il costituente principale dello scheletro, nella frazione sabbiosa e limosa prevalgono le particelle monominerarie. L’espansione termica differenziale dei diversi minerali è un processo pedogenetico importante nel determinare la disaggregazione delle rocce e la formazione colluvium su cui è impostato il suolo. L’ordine di alterabilità dei minerali osservati rispecchia quello previsto su basi termodinamiche ed è (Faure G., 1992):
solfuri > serpentino > feldspati > miche >
quarzo
I solfuri risultano completamente alterati in limonite. Processi di alterazione chimica sono evidenti sulle superfici dei del serpentino e dei feldspati anche quando questi hanno le dimensioni del limo. Sulla superficie delle miche, minerali considerati tra i meno alterabili, sono evidenti solo processi di esfoliazione meccanica. Il quarzo non evidenzia processi di alterazione chimica o meccanica eccetto che la frammentazione determinata dagli urti meccanici.
Le particelle minerali sono cementate da composti organici mucillaginosi e filamentosi. Negli aggregati irregolari le particelle minerali sono completamente immerse in una matrice costituita da sostanza organica. Questa, presumibilmente costituita da essudati radicali e composti umici, penetra tra i granuli minerali e inglobandoli completamente. La sostanza organica ha quindi una grande importanza nel determinare lo stato di aggregazione dei osservata nei profili pedologici, la macroporosità, la bagnabilità, e quindi la porosità del suolo e l’infiltrazione efficace delle piogge. La
Figura 426-1.
Figura 426-2.
porosità e l’infiltrazione efficace sono proprietà del suolo che condizionano la frazione di acque piovane che si infiltrano nel suolo e la loro velocità di discesa verso la falda e, in ultima analisi, la velocità di trasporto degli inquinanti.
4.2.6. Composizione
mineralogica dei suoli di Villadossola.
Nel corso del rilevamento della composizione mineralogica delle argille presenti nei primi 15 cm dei suoli Piemontesi condotta da Facchinelli et al. (1997), due suoli raccolti sono stati raccolti nell’area in esame: il Campione “Domodossola”, sulle alluvioni del fiume Toce, ed il Campione “Pontetto”, su un terrazzo glaciale. I diffrattogrammi e le stime semiquantitative (F426-1, F426-2) indicano che la vermiculite rappresenta rispettivamente il 20 e il 30% della frazione argillosa, mentre non risulta rappresentata la Smectite, peraltro alquanto rara nella regione e rappresentata nell’arco alpino principalmente nei suoli sviluppati sui complessi serpentinitici. Nelle argille della spalla glaciale, (F426-1) la scomparsa del picco a 18Å del campione saturato in Mg a seguito del trattamento Co K indica la presenza di interstratificati Vermiculite-Smectite.
L’elevato contenuto di vermiculite osservato nelle argille delle spalle glaciali è in buon accordo con l’elevata capacità di scambio cationico della frazione minerale di questa unità geomorgfologica.
4.3.1. Stima della
concentrazione litogenica ed antropica.
La concentrazione dell’elemento di transizione calcolato dall’inventario geochimico e la concentrazione media dell’elemento estraibile in acquaregia sono riportate nella tabelle T431-1 e T431-2. Nel profilo di Cliland si osserva che i metalli pesanti estratti dall’acquaregia hanno una concentrazione assai prossima a quella dell’inventario geochimico eccetto che per Cd e Pb. Nei suoli di Villadossola si osservano concentrazioni a quelle stimate dall’inventario geochimico per tutti gli elementi eccetto che per Co, Ni e Cr. La distribuzione dei metalli pesanti è diversa nei suoli disturbati dalle lavorazioni e nei suoli non disturbati dopo l’evento inquinante del 1980-1982. Nei suoli non disturbati dalle lavorazioni si osserva che la concentrazione di tutti gli elementi in traccia eccetto quelle di Co e Ni decresce esponenzialmente con la profondità fino a raggiungere valori prossimi a quelli stimati dall’inventario geochimico a profondità superiori ai 20 cm (Tabella T431-2). Nel confrontare la concentrazione litogenica osservata e quella stimata, va osservato che l’acquaregia solubilizza
Tabella T431-1.
Inventario geochimico della Val d’Ossola
|
Roccia |
|
Pb |
Cu |
Ni |
Zn |
As |
Bi |
Cr |
Sb |
Cd |
Co |
|
|
% |
Concentrazione, mg/kg (*) |
|||||||||
|
Gneiss |
80.5 |
21 |
12 |
3 |
51 |
1 |
0 |
2 |
0 |
0 |
1 |
|
Rocce
carbonatiche |
10.9 |
15 |
25 |
47 |
62 |
12 |
1 |
40 |
1 |
0 |
10 |
|
Anfiboliti |
4.6 |
5 |
43 |
150 |
97 |
2 |
0 |
245 |
0 |
0 |
43 |
|
Quarziti |
1.9 |
15 |
11 |
2 |
40 |
1 |
0 |
30 |
0 |
0 |
1 |
|
Serpentiniti |
1.7 |
1 |
35 |
2000 |
58 |
3 |
1 |
3200 |
0 |
0 |
155 |
|
Gneiss
anfibolitici |
0.7 |
13 |
50 |
76 |
74 |
1 |
0 |
230 |
0 |
0 |
22 |
|
|
|
Concentrazione ponderata con la superficie
di affioramento, mg/kg. |
|||||||||
|
Gneiss |
80.5 |
16.91 |
9.66 |
2.01 |
41.06 |
0.81 |
0.10 |
1.61 |
0.08 |
0.11 |
0.81 |
|
Rocce
carbonatiche |
10.9 |
1.60 |
2.67 |
5.12 |
6.70 |
1.31 |
0.08 |
4.36 |
0.05 |
0.03 |
1.04 |
|
Anfiboliti |
4.6 |
0.23 |
1.98 |
6.90 |
4.46 |
0.07 |
0.00 |
11.27 |
0.00 |
0.01 |
1.98 |
|
Quarziti |
1.9 |
0.29 |
0.21 |
0.04 |
0.76 |
0.02 |
0.00 |
0.57 |
0.00 |
0.00 |
0.01 |
|
Serpentiniti |
1.7 |
0.02 |
0.60 |
34.00 |
0.99 |
0.05 |
0.02 |
54.40 |
0.00 |
0.00 |
2.64 |
|
Gneiss
anfibolitici |
0.7 |
0.09 |
0.35 |
0.53 |
0.52 |
0.01 |
0.00 |
1.61 |
0.00 |
0.00 |
0.15 |
|
Concentrazione
litogenetica del suolo |
19.1 |
15.5 |
48.6 |
54.5 |
2.3 |
0.20 |
73.8 |
0.14 |
0.15 |
6.6 |
|
(*) La
concentrazione media degli elementi in traccia nelle rocce è elaborata dai dati
riportati in: Wedepohl K.H., 1978,
"Handbook of Geochemistry".
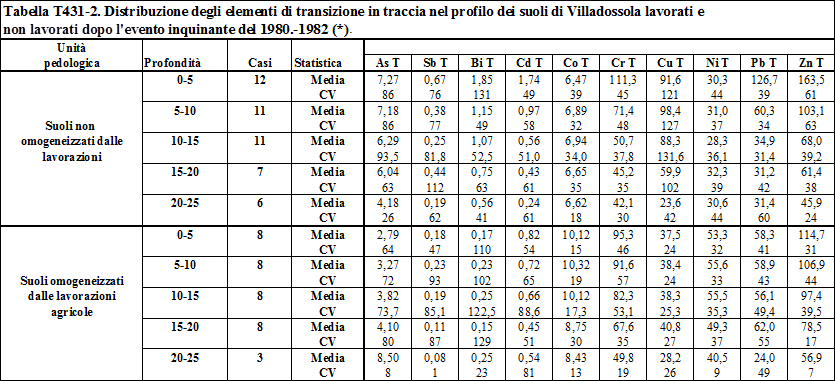
l’elemento legato alla sostanza organica, ai sesquiossidi, ai solfuri, e solo parzialmente o poco l’elemento occluso nei silicati primari e nelle Vermiculiti (Page L. et al. Eds, 1982). La concentrazione dell’elemento stimata dall’inventario geochimico, esprime la concentrazione dell’elemento totale e comprende quello occluso nei minerali primari, che come è stato osservato nello studio dei cicli petrogenici degli elementi, non è trascurabile.
Per stimare il contenuto litogenico si è proceduto diversamente nei suoli di Villadossola e nel suolo di Cliland. Nei suoli di Villadossola, che si sviluppano su un terreno geologico attraversato da importanti mineralizzazione a solfuri, è stato assunto che la concentrazione litogenica sia il valore minore tra la concentrazione stimata dall’inventario geochimico e la concentrazione osservata negli strati più profondi dei profili. Nel profilo di Cliland, che si sviluppa su un terreno geologico poverissimo di mineralizzazioni (Cameron B.I. e Stephenson D., 1985) si è assunto, seguendo Page L.
et al. Eds., 1982) l’elemento estratto dall’acquregia sia composto principalmente dall’elemento antropico mente l’elemento litogenico sia per lo più in forme non dissolubili da un attacco acido forte. La stima della concentrazione dell’elemento litogenico ed antropico è arbitraria in entrambe le aree, ma va osservato che, come emerge dall’esame critico della letteratura discusso nel paragrafo 3.6 del capitolo dedicato ai materiali e ai metodi, eccetto che quando sia possibile effettuare analisi isotopiche, non vi sono metodi considerati universalmente validi e comunemente accettati che permettano di determinare la concentrazione litogenica dell’elemento.
L’arricchimento antropico di un elemento nel suolo (AA) è definito come il rapporto tra la concentrazione dell’elemento totale osservata nel suolo e la concentrazione dell’elemento litogenico (tabella T432-1). Esso è un parametro utile per valutare la perturbazione dei cicli biogeochimici, in quanto esattamente come il flusso dell’elemento che dal suolo entra nella biomassa vegetale (Sillampää M., 1982. Baes C.F. et al., 1984) i flussi dei cicli biogeochimici dei metalli pesanti sono per lo più governati da cinetiche lineari (Lasaga A. C, 1980. Whitfield M., 1981. Whitfield M., e Turner D.R., 1982. Mckenzie et al., 1979). Nel suolo di Cliland e nei suoli di Villadossola, si osserva che l’arricchimento antropico segue lo stesso ordine, e che il fattore di arricchimento antropico di Co, Ni, Zn, Cd, Pb e Cr del suolo inquinato dalle attività siderurgiche è correlato al fattore di arricchimento dell’elemento nel suolo rispetto alla roccia madre (FAS) osservato in Nord America (Shacklette H. T. et al., 1984), tabella T432-1 e figura F432-1):
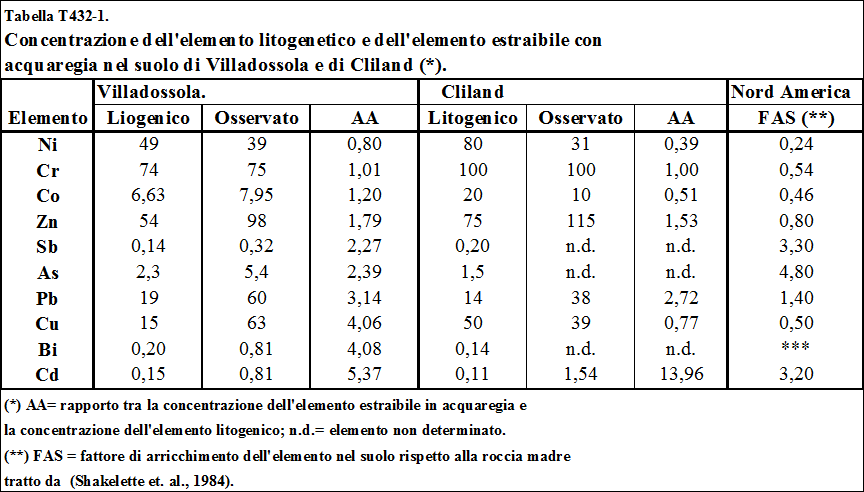
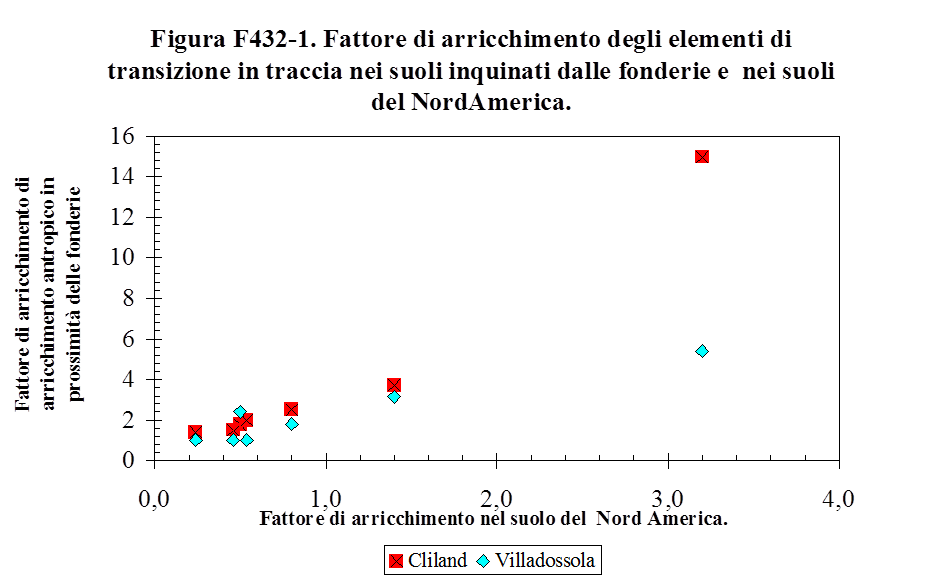
FAS =
(0,031 +/- 0,97) + (0,31 +/- 0,023) * AA
n = 7; r2 = 0,997; P=7,6*10^-5
La stretta relazione osservata tra arricchimento antropico del suolo contaminato dalle emissioni delle fonderie e il fattore di arricchimento di Co, Ni, Zn, Cd, Pb e Cr rispetto alla roccia madre osservato nel Nord America lascia supporre che il suolo del Nord America sia inquinato dalle emissioni delle fonderie. Il contenuto fattore di arricchimento dell’As nel suolo del Nord America è molto superiore a quello stimabile dall’inquinamento siderurgico e può essere spiegato dal massiccio uso dell’As come pesticida e defoliante (Alloway D.C., 1986). L’arricchimento di Sb nel suolo del Nord America è un poco inferiore a quello prevedibile dall’arricchimento di Co, Ni, Zn, Cd, Pb e Cr, ma segue lo stesso andamento. Non si hanno dati sull’arricchimento del Bi nel suolo del Nord America, ma questo può essere stimato di circa 1,3.
Nei suoli di Villadossola l’arricchimento antropico è ovviamente diverso attraverso il profilo dei suoli che sono stati lavorati a seguito dell’evento inquinante del biennio 1986-1986 ed in quelli che, dopo aver ricevuto l’immissione siderurgica, sono stati lavorati. I valori più elevati dei fattori di arricchimento antropico si registrano ovviamente nello strato superficiale dei suoli indisturbati, dove sono in ordine decrescente 11 (Cd), 10 (Bi), 5.9 (Cu), 6.6 (Pb), 3.6 (Zn), 3.3 (Sb), 3.2 (As) e 2.6 (Cr). Nei suoli lavorati i fattori di arricchimento sono ovviamente inferiori ed approssimativamente costanti attraverso il profilo. I valori medi decrescono nell’ordine 4.2 (Cd), 2.7 (Pb), 2.4 (Bi), 2.4 (Cu), 2.0 (As), 2.0 (Zn), 1.8 (Cr).
La lisciviazione degli inquinanti è stata indagata mediante un modello matematico che esprime la mobilità dell’inquinante attraverso tre diversi parametri: la velocità di lisciviazione, la costante cinetica dell’inquinante da un volume di suolo al sottostante e la costante di ripartizione in situ. I tre parametri forniscono informazioni complementari e differenti sui meccanismi che controllano la velocità di lisciviazione verso la falda idrica. Le principali proprietà chimiche e fisiche dei suoli di Villadossola su cui si è studiata la lisciviazione sono riportate nell’appendice A6. Le velocità di lisciviazione ed i tempi di residenza calcolati sono riportati nelle appendice A7.
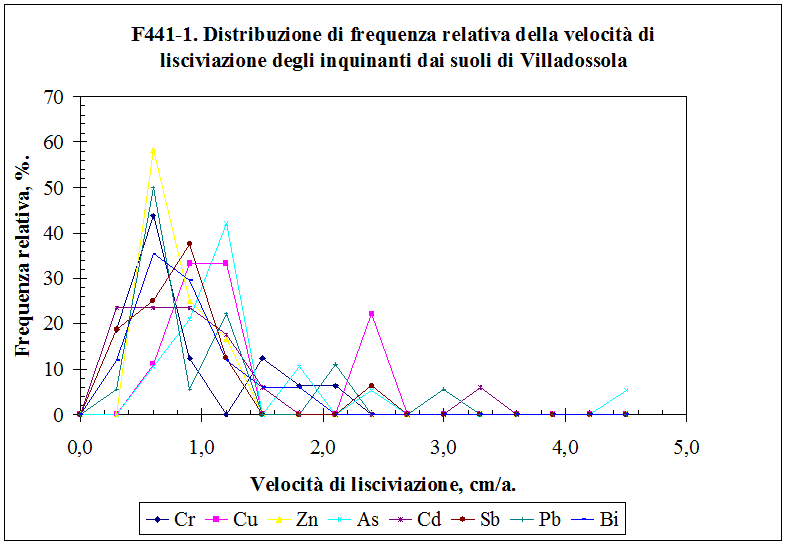 |
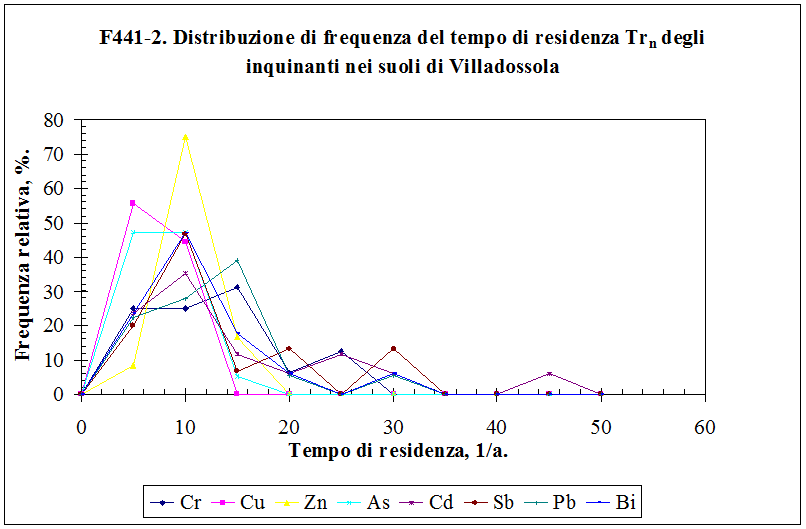
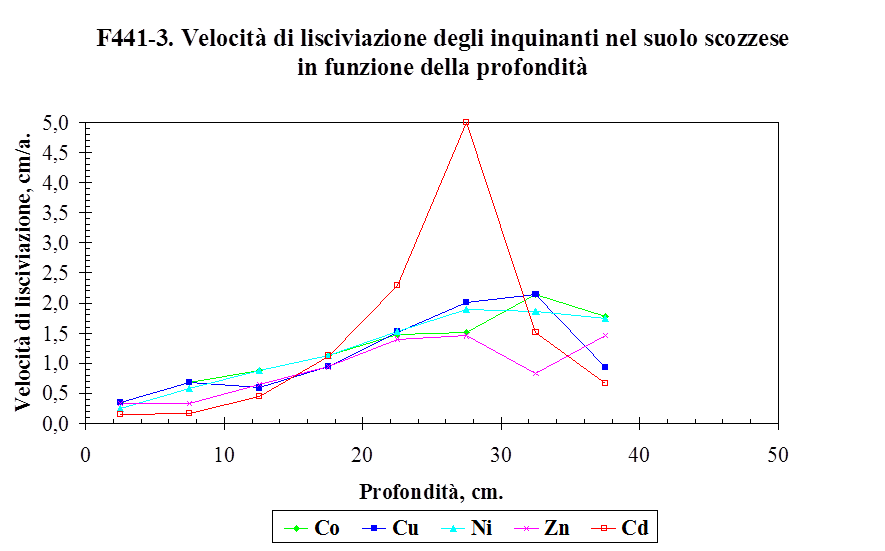
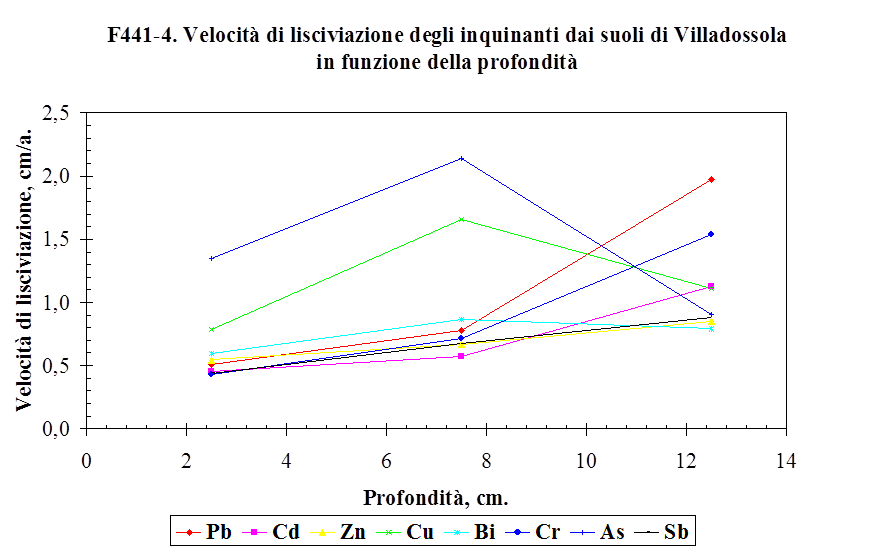



4.4.1. La velocità di lisciviazione.
Le velocità di lisciviazione medie (cm/a) hanno una deviazione standard compresa tra il 30 ed il 140% (figura F441-1 e F441-2) e sono in ordine decrescente:
a Cliland
Cd(1,4)>Co(1,2)=Ni(1,2)>Cu(1,1)>Pb(0,9)>Zn
(0,9)
a Villadossola
As (1,4)>Cu
(1,2)>Pb(0,8)>Cd(0,75)=Cr(0,74)=Sb(0,7)Bi>(0.78)>Zn (0,66)
Nel profilo di Cliland la velocità di lisciviazione degli inquinanti, tutti elementi di transizione bivalenti, è minima nello strato superficiale, aumenta con la profondità raggiungendo il valore massimo nell’orizzonte Ae per tornare a diminuire lievemente in prossimità dell’orizzonte Bg (F441-3). Particolarmente marcato è l’aumento della velocità di lisciviazione del Cd in corrispondenza dell’orizzonte Ae. Nei suoli di Villadossola la velocità di lisciviazione di Pb, Cd, Zn, Sb, e Cr aumenta muovendo dagli orizzonti superficiali a quelli profondi (F441-4). Dal comportamento medio si discosta il Cu, elemento che può essere pervenuto al suolo a causa della coltura della vite e dell’As, un elemento che può essere metabolizzato dalle popolazioni fungine e batteriche in forme metilate volatili (Tamaki S. e Frankerbergher W. T., 1992). Le regressioni multiple significative ottenute con il metodo della progressiva aggiunta di variabili tra velocità di lisciviazione e proprietà chimiche e fisiche del suolo sono riportate nella tabella T441-1. Nel profilo di Cliland si osserva che la velocità di lisciviazione è controllata dal contenuto in sostanza organica e dalla capacità di scambio cationico. Per i metalli a basso potenziale ionico la proprietà del suolo che meglio spiega la velocità di lisciviazione è il contenuto in sostanza organica, mentre per i metalli a basso potenziale ionico la proprietà che meglio spiega la velocità di lisciviazione è la capacità di scambio cationico, confermando il criterio generale secondo cui nelle serie litotropiche l’importanza delle argille nel legare il metallo aumenta al diminuire del potenziale ionico (Violante P., 1986b). Nei suoli di Villadossola si osserva che la sostanza organica rallenta la lisciviazione dell’inquinante, mentre la frazione minerale, sia essa sabbia fine o frazione fine limo+argilla è positivamente correlata alla velocità di lisciviazione. Si osserva inoltre che anche la densità del suolo è positivamente correlata alla velocità di lisciviazione. L’analisi statistica dei dati conferma quanto osservato dalla distribuzione delle velocità di lisciviazione attraverso il profilo del suolo, ovvero un aumento della velocità passando dall’orizzonte superficiale ricco in sostanza organica e poroso agli orizzonti sottostanti minerali e densi. L’osservazione che la frazione fine è negativamente correlata alla velocità di lisciviazione suggerisce che la frazione di metallo pesante mobile sia un complesso anionico.
4.4.3. Il tempo di residenza
dell’inquinante nel suolo.
Come risulta dalle regressioni multiple ottenuto con il metodo della progressiva aggiunta di variabile, il tempo di residenza degli inquinanti nel suolo di Villadossola è controllato dal contenuto di sostanza organica e dalla tessitura (tabella T442-1). Il tempo di residenza aumenta con il contenuto di sostanza organica e diminuisce sia con il contenuto di sabbia fine che di frazione fine limo + argilla. Per gli elementi per i quali la tessitura non influisce significativamente nel determinare il tempo di residenza nel suolo (Cu, Zn, Cd), il tempo di residenza può essere efficacemente espresso da un modello lineare che trascura l’influenza della sostanza organica e può essere espresso con una singolare precisione dalla formula:
Una relazione simile si osserva nel profilo di Cliland (figura F442-1). Sia nei suoli di Villadossola che nel suolo di Cliland tra costante A e potenziale ionico PI dell’elemento sussiste una stretta relazione:
Villadossola,
A = -(2,13+/-0,034) + (9,50+/-0,08)/PI; n=3; r2=0.999; P=0,005
Cliland,
A = -(2,08+/-1,01) + (10,10+/-2.44)/PI; n=6; r2=0.810; P=0.015
La relazione tra costante A e potenziale ionico dell’elemento è simile nelle due aree in esame, ed i coefficienti della regressione differiscono meno del 10%. La relazione tra la costante A ed il tempo di residenza è molto simile alla relazione che intercorre la costante di stabilità K2 del complesso binario con l’acido acetico M(Ac)2 ed il potenziale ionico dell’elemento:
Ks2 = (1,6+/-0,4) + (9,5+/-0,8)/PI; n=6; r2=0,987;
P=0,05

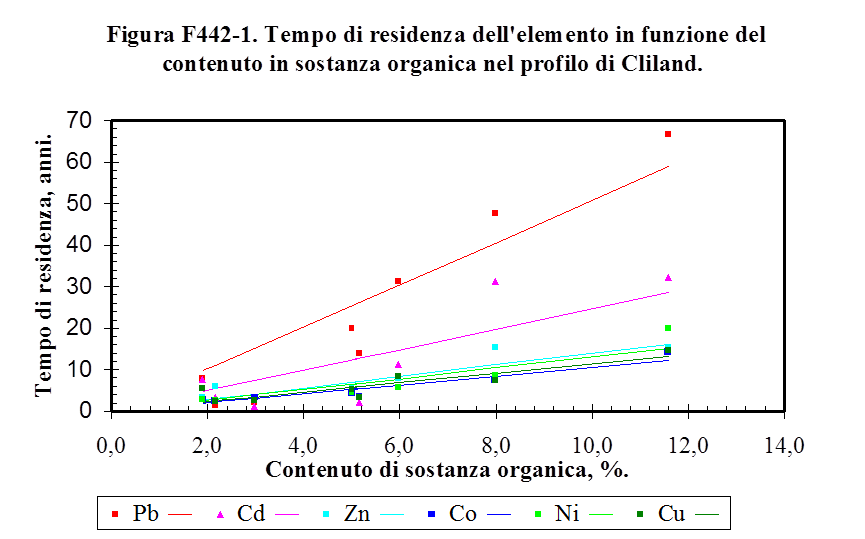
Considerato che tra tempo di residenza Trn dell’inquinante nello strato di suolo n e la costante cinetica del rilascio Kn dell’inquinante dallo strato n al sottostante vale la relazione:
Trn = 1/Kn
per gli elementi di transizione bivalenti la costante cinetica della lisciviazione può essere espressa con dalla formula:
Kn = [K2 * SO]-1
dove:
K2
= è la costante di stabililità del complesso (CH3COO)2M
SO = è la concentrazione della sostanza organica nel suolo
La relazione riportata predice la cinetica della lisciviazione da uno strato di suolo al sottostante di Co, Ni, Cu, Ni, Cd e Pb con un’incertezza del 10% indipendentemente dalla tessitura tutti i suoli osservati dove il pH è compreso tra 4 e 5 ed il contenuto di sostanza organica supera il 3g/100.
La relazione osservata tra cinetica del rilascio dell’inquinante cationico bivalente e stabilità del complesso binario con l’acido acetico è in buon accordo con le indagini di laboratorio dalle quali risulta che la affinità dei gruppi carbossilici degli acidi umici per i metalli di transizione bivalenti è prossima a quella dei gruppi carbossilici dell’acido acetico (Munza B. et al., 1995). L’osservazione è inoltre in accordo con i risultati delle indagini in vitro dalle quali risulta che nell’intervallo di pH compreso tra 4 e 5, quello prevalente nei suoli indagati, il complesso che i composti umici formano con i metalli di transizione bivalenti è quello binario M-(OOC-R)2 (Shnitzer M, 1991).
Il modello a serbatoi e flussi ben descrive la mobilità verticale degli inquinanti e permette di identificare le proprietà chimico-fisiche che la condizionano. Esso permette inoltre di stimare il tempo necessario perché gli inquinanti vengano lisciviati al di sotto dello strato in cui l’attività radicale è più intensa.
F432-1.
Le relazioni osservate tra le proprietà del suolo e le costanti cinetiche del rilascio dell’inquinante da uno strato al sottostante permettono ovviamente una stima più precisa dei tempi di autodepurazione del suolo. Nella figura F423-1 è rappresentata la lisciviazione del Cu da un suolo avente le proprietà chimico fisiche medie dei suoli di Villadossola. Si può osservare che la distribuzione dell’inquinante attraverso il profilo evolve gradualmente da una forma esponenziale ad una curva gaussiana allargata. Simulazioni simili effettuate per gli altri elementi permettono di stimare che il tempo medio necessario affinché gli inquinanti raggiungano nei primi 15 cm di suolo concentrazioni prossime a quelle litogeniche sono compresi tra i 30 ed i 60 anni in funzione dell’elemento e delle proprietà del suolo. Si può comunque prevedere che in tale lasso di tempo la quasi totalità degli inquinanti non raggiunga profondità superiori al mezzo metro, rimando così biodisponibili per le specie arboree e arbustive per un lasso di tempo nell’ordine di 1-2 secoli.
La concentrazione di Co, Ni, Cu, Zn, Cd e Pb estraibili dal reagente di Lakanen è riportata nell’appendice A5. I valori medi dell’elemento biodisponibile espressi in mg/Kg sono riportati nella tabella T45-1. Nell’insieme dei suoli campionati la concentrazione dell’elemento biodisponibile (mg/Kg) è sempre correlata all’elemento totale con un limite di confidenza superiore al 5% (T45-2). La frazione biodisponibile percentuale media degli elementi è in ordine decrescente:
Co (3+/-38%)<Ni
(5+/-15%)<Cd (35+/-14%)<Zn (57+/-5%)<Cu (68+/-2%)< Pb (79+/-5%)
Gli inquinanti (Pb, Cd, Zn, Cu) sono caratterizzati dall’essere biodisponibili in una quota percentuale superiore di un ordine di grandezza a quella degli elementi di origine prevalentemente litogenica (Co, Ni). L’incertezza con la quale la concentrazione dell’elemento inquinante biodisponibile può essere prevista dalla concentrazione totale è compresa tra il 2 ed il 14%. Per gli elementi litogenici l’incertezza con la quale la concentrazione dell’elemento biodisponibile è prevedibile dall’estrazione con l’acqua regia è superiore a quella degli inquinanti, ed è compresa tra il 15 ed il 38%.
Nei suoli che a seguito dell’evento inquinante del biennio 1986-7 sono stati rimaneggiati dalle lavorazioni e quindi destinati a prato permanente la frazione biodisponibile di Pb, Cd e Zn è controllata dalla frazione fine limo + argilla e dalla sostanza organica:
Zn Fb = 0.194 + 0.0263 ·S.O. -0.0150 ·F; P = 5.7 ·10-7; n=30; r = 0.809
Cd Fb = 0.864 + 0.0482
·S.O. - 0.0463 ·F; P = 2.0 ·10-6; n=30; r = 0.788
Pb Fb = 0.306 + 0.0337
·S.O. -0.00414 ·F; P = 6.4 ·10-3; n=30; r = 0.558

Una analoga relazione si osserva quando si considera tutto l’insieme dei suoli
campionati:
Pb Fb = 0,455 + 0,1300*S.O. -
0,0097*F; r2=0,256;
n= 77; P>0,001
Cd Fb = 0,688 + 0,0130*S.O. - 0,0295*F; r2=0,428; n=
77; P<0,001
Zn Fb = 0,205 + 0,0130*S.O. - 0,0109*F; r2=0,609; n=
77; P<0,001
Cu Fb = 0,354 + 0,0452*S.O. - 0,0049*F; r2=0,609; n= 77; P<0,001
Le relazioni fra sostanza organica e frazione minerale suggeriscono che la frazione biodisponibile degli inquinanti sia controllata dalla reazione di scambio:
Argilla-M +
2HOOC-R = Argilla-H + M(OOC-R)2
La frazione tessiturale che controlla la biodisponibilità è diversa in funzione della unità geomorfologica. Nelle spalle glaciali e nei suoli sviluppati sulle alluvioni del fondovalle la frazione biodisponibile dell’inquinante è negativamente correlata alla frazione fine del suolo, mentre sugli acclivi versanti vallivi è negativamente correlata alla sabbia fine (tabella T45-2). L’osservazione è in buon accordo con quanto osservato al riguardo della capacità di scambio cationico della frazione minerale, che risulta determinata dalla frazione fine nelle aree pianeggianti ed è controllata dalla sabbia fine sugli acclivi versanti vallivi.
L’osservazione che la capacità di scambio cationico della frazione minerale del suolo lega l’inquinante con legami forti bloccandolo in forme non biodisponibili è in contraddizione con l’osservazione che la velocità di lisciviazione è inversamente proporzionale al contenuto di frazione fine limo + argilla. E’ noto che le Illiti dei suoli attorno al nucleo di mica inalterato presentano dei bordi di alterazione in vermiculite (Sawhney B.L., 1972. Bolt G.H. et al., 1963. Brouwer E. et al., 1983. Cremers A. et al., 1988. Francis C. W. e Brincley F. S., 1976. Grutter A. et al., 1986. Hill D. E. e Sawhney B. L., 1969. Jackson M. L., 1963. Klobe W. D. and Gast R. G., 1970. Le Roux J. e Rich C.I., 1969. Le Roux J. et al., 1970. Rich C.I. e Black W. R., 1964. Sawhney B.L., 1969. Sawhney B.L., 1970). Le Illiti possono legare i cationi a bassa energia di idratazione con tre diversi siti, i silossilanici posti sulle superfici esterne dei cristalli, i siti silossilanici intrareticolari posti negli interstrati a spaziatura di 14Ä dell’orlo esterno del cristallo alterato in vermiculite e nei siti di cuneo interno che fanno da transizione tra il nucleo cristallino interno e il domino cristallino esterno alterato. Le isoterme di adsorbimento (Bolth G.H., 1979) indicano che i primi due siti hanno un’energia di

legame nei confronti degli metalli di transizione molto inferiore a quella formata con i composti umici. I siti di cuneo interno possono al contrario formare legame con cationi caratterizzati da basse energia di idratazione che hanno un ordine di grandezza confrontabile a quello dei composti ionici. I siti di cuneo
interno possono quindi competere con l’EDTA del reagente di Lakanen nel determinare la concentrazione del metallo presente nell’estratto. I composti umici sono adsorbiti sui minerali delle argille in corrispondenza dei bordi del cristallo legandosi attraverso i gruppi carbossilici ai metalli dello strato ottaedrico (Schnitzer M., 1991. Koyama M., 1995). Inoltre nell’interstrato delle vermiculiti possono penetrare composti molecole organiche quali i composti alifatici contenenti fino a 12 molecole di carbonio purché contenenti un radicale amminico (Wilson M.J., 1987) Recenti ricerche rese pubbliche in occasione del congresso della Società Italiana di Scienza del Suolo del 1999 indicano che incubando le Illiti con sostanza organica questa può ostruire i siti di cuneo interno impedendone la reazione con il catione a bassa energia di idratazione ed impedendone la fissazione sul complesso di scambio. I dati riportati in letteratura sull’adsorbimento dei cationi a bassa energia di idratazione e le osservazioni condotte nella presente ricerca suggeriscono che la attività biologica e la sostanza organica del suolo impedisca ai siti di bordo interno ubicati sui bordi di alterazioni di fissare i cationi a bassa energia di idratazione, così che sembra ragionevole dubitare che in condizioni di terreno la solubilità e la biodisponibilità dell’inquinante nei suoli considerati sia principalmente determinata dai complessi organometallici.
4.6. Distribuzione areale di As, Bi, Sb, Co, Ni, Zn, Cu, Cd
e Pb nei suoli del comune di Villadossola.
Nella appendice A8 è riportata la quantità di metallo pesante presente nei primi 15 cm dei suoli indisturbati riferita ad un m2. Per i suoli che dopo l’evento inquinante sono stati omogeneizzati dalle lavorazioni la quantità di metallo è stata integrata attraverso l’intero spessore dell’orizzonte lavorato. I colori verde, giallo, arancione e rosso si riferiscono rispettivo al primo, secondo, terzo e quarto quartile. Per tutti gli elementi considerati, Il carico di metallo pesante per unità di superficie differisce dal primo al quarto quartile di un ordine di grandezza. E’ evidente dalle figure che la distribuzione attorno allo stabilimento siderurgico è alquanto irregolare e non lascia evincere una chiarissima distribuzione spaziale. In linea generale, si può osservare che l’area rossa, relativa all carico di metallo pesante maggiore, si allunga un circa uno-due chilometri sottovento allo stabilimento siderurgico e lascia luogo a campioni gialli o arancione. Sul versante destro della valle, prevalgono i campioni gialli o arancione. Sul versante sinistro della valle prevale di norma il colore verde, sebbene siano talvolta presenti campioni gialli.
4.7. Raffronto delle concentrazioni totale e biodisponibile con i limiti di legge.
Nessuno degli elementi considerati supera i limiti di soglia posti dalla normativa regionale per gli usi industriali. Per quanto concerne il Cu, si osserva che il limite di legge per l’uso agricolo e residenziale viene abbondantemente superato nei suoli dove è stata coltivata la vite. Nei suoli indisturbati che non hanno in passato la coltivazione della vite la concentrazione del Cu è sempre inferiore ai limiti di legge vigenti. I limiti di legge per l’uso residenziale e agricolo vengono per contro approssimati o superati dalle concentrazioni di Pb, Zn, Cu, e Cd, rispettivamente nel 2, 5, 9 e 10% della popolazione campionaria. Le soglie di bonifica di Pb e Cd sono superate nei suoli lavorati entro un raggio di 1-2 Km sottovento allo stabilimento siderurgico e negli strati più superficiali dei suoli indisturbati in tutta l’estensione dell’area studiata.
Tra gli elementi considerati a causa dell’elevata tossicità merita particolare attenzione il Cd. Per alcune specie quali la lattuga e lo spinacio si osserva una diminuzione del 25% della biomassa sostenuta dal suolo a soli 2-4 mg/kg di elemento pseudototale (Alloway D.C, 1986). In prossimità dello stabilimento siderurgico e in suoli dotati di 2 mg/kg di Cd totale alcuni agricoltori lamentano l’ingiallimento delle specie foraggiere. Il Cd infine si concentra in talune specie orticole quali la lattuga e lo spinacio e può dar luogo a fenomeni di accumulo nella biomassa.
5.
Il livello di inquinamento dei suoli è elevato in entrambe le aree studiate. Il fattore di arricchimento antropico, ovvero il rapporto tra l’elemento antropico e quello litogenico, stimato sulla base degli inventari geochimici segue lo stesso ordine in Scozia e a Villadossola:
Cliland Cd(14.96)>Pb(3.72) > Zn(2.53) >
Cr(2,00) > Cu(1,77) >Co (1.51) > Ni(1.39)
Villadossola Cd(
5.45)>Pb(3,05)>Zn(2,.50)>Cu(2,43)>Cr(2,26) >Ni(1,75) >
Co(1.53)
I fattori di arricchimento antropici stimati in Italia ed in Scozia sono fortemente correlati (r2 > 0,960, P<0,001) ai fattori di arricchimento suolo/roccia madre osservati nel Nord America a conferman delle ipotesi che le attività siderurgiche abbiano determinato nell’emisfero australe un forte aumento della concentrazione nel suolo di Cd, Pb, Zn, Cu e Cr.
L’elemento biodisponibile determinato nei suoli di Villadossola è ben correlato all’elemento estraibile in acqua regia (r2>0,85) ed è nell’ordine (%):
Villadossola: Pb 79 > Cu 68 > Zn 57 > Cd 35 >
Ni 5 > Co 3.
Indicando che nei suoli in esame i fattori di trasferimento fornicono una stima relativamente accurata della concentrazione degli inquinanti nei veegetali.
La frazione biodisponibile di Cd, Pb, Zn, Cu è determinata dalla sostanza organica e dalle argille presenti nella frazione fine F (limo+ argilla):
Pb fb = 0,455 +
0,1300*S.O. - 0,0097*F; r2=0,256;
n= 77; P>0,001
Cd fb = 0,688 + 0,0130*S.O. -
0,0295*F; r2=0,428; n= 77;
P<0,001
Zn fb = 0,205 + 0,0130*S.O. -
0,0109*F; r2=0,609; n= 77;
P<0,001
Cu fb = 0,354 + 0,0452*S.O. - 0,0049*F; r2=0,609; n= 77; P<0,001
La frazione fine blocca l’elemento in forme non biodisponibili mentre la sostanza organica lo complessa in forme biodisponibili. All’aumentare della costante di stabilità dei complessi organometallici aumenta l’importanza della sostanza organica e diminuisce quella delle argille. La biodisponibilità dell’elemento è quindi controllata dalla reazione
Argilla-M +
(HOOC-R)2 =
Argilla-H + M-(OOC-R)2
il cui equilibrio è spostato a destra per gli elementi che come il Cu e Pb formano stabili complessi organometallici.
La velocità di lisciviazione media degli inquinanti verso la falda è nell’ordine (cm/a):
Cliland: Cd(1,4)>Co
(1,24)> Ni(1,23)>Cu(1,14)>Pb(0,92)>Zn(0,92)
Villadossola: As(1,42)>Cu(1,16)>Pb(0,83)>Cd(0,75)>Cr(0,74)>Sb(0,70)>Zn(0,66)
Nel profilo di Cliland, dove la tessitura del suolo è relativamente omogenea, la velocità di lisciviazione è principalmente limitata dal contenuto di sostanza per i metalli di transizione bivalenti ad elevato potenziale ionico e dalla capacità di scambio cationico per Cd e Pb. Nei suoli di Villadossola si osserva che la velocità di lisciviazione degli elementi presenti n forma cationica e anionica è controllata, oltre che dal contenuto in sostanza organica (SO), dalla sabbia fine (SF):
V Pb =0,244 – 6,70*SO + 1,9*SF; n=18; r2=0,639;
P<0,001
V Cd
= 1,64 – 16,2*SO – 0,132*SF ; n=17; r2=0,264 ; P=0,16
V Zn =
0,529 – 2,92*SO + 0,432*SF; n=12 ; r2=0,546 ;
P=0,029
V Sb = -0,278 + 4,92*SO + 1,15*SF; n=16; r2=0.531
; P=0.007
V Cr = -0,784 - 11,3*SO + 8,45*SF; n=16 ; r2=0.678 ; P<0,001
V Cu = 5,78 – 5.83*SO + 1,36*SF; n=10; r2=0,605; P=0,0039
La tessitura sciolta favorisce lo sgrondo delle acque e la lisciviazione degli inquinanti, mentre la sostanza organica li complessa in forme biodisponibili e poco mobili. La frazione fine non ritiene gli inquinanti presumibilmente sia poiché è completamente inglobata dai colloidi organici, sia poiché le forme più mobili sono i complessi datati di carica negativa. L’applicazione del modello a serbatoi e flussi permette di stimare che il tempo richiesto affinché gli inquinanti raggiungano le concentrazioni litogeniche “normali” nei primi 15 cm di suolo, dove l’attività radicale delle piante erbacee è maggiore, in dipendenza dell’elemento e delle proprietà del suolo, è compreso tra 30 e 60 anni. Il tempo necessario affinché gli inquinanti raggiungano concentrazioni le litogeniche nel primo metro di suolo, è compreso tra 50 e 100 anni.
La dispersione degli inquinanti attorno allo stabilimento è controllata dalla direzione dei venti. Le concentrazioni di inquinanti più elevate si osservano sottovento lo stabilimento siderurgico entro una distanza di 1-2 Km, mentre concentrazioni moderatamente elevate si estendono sottovento a distanze superiori ai 7 Km.
6.
BIBLIOGRAFIA.
Adriano D.C., 1986. “Trace elements in the terrestrial environment”. Springer-Verlag, Berlin.
Ajmone Marsan F., Bain D.C., and Duthie D.M.L., 1988. Parent material uniformity and degree of weathering in soil chronosequence, northwestern Italy”, Catena, 15: 507-517.
Alloway B.J Ed.., 1995. “Heavy Metals in soils”. Blackie Academic and Professional, London
Al-Saleh Iman, and Coate Laura, 1995. Lead exposure in Saudi Arabia from the use of traditional cosmetics and medical remedies. Environmental Geochemistry and Health, 17: 29-31.
Aleksandrova L.N., 1967. Organomineral humic acid derivates and method of studing them. Soviet Soil Science, 7: 903-913.
Alfy S. El, and Rassoul-Abdel A.A., 1993. Trace metal pollutants in el manzala lakes by inductevely coupled plasma spectroscopy. Water Research, 7: 1253-1256.
Allison L.E., 1960. Wet combustion apparatus and procedure for organic and inorganic carbon in soil. Soil Science Society American Proceedings, 24:36-40.
Anderson M.P., 1979. Using models to simulate the movement of contaminants through groundwater flow systems. Critical Review on Environtal Control, 9: 97-156.
Andrew J.E., Brimblecombe P., Jickells T.D., and Liss P.S., 1996. “An introduction to environmental chemistry”. Blackwell Science.
Aoyama Masakazu, and Itaya Seiji, 1995. Effects of copper on the metabolism of 14C-labeled glucose in soil in relation to amendment with organic materials. Soil Science and Plant Nutrition, 41: 245-252.
Ardakani M.S., and Stevenson F.J., 1972. A modified ion-exchange techniques for the determanation of stability constant of metal-soil organic matter complexes. Soil Science Society of America Proceedings, 36: 884-890.
Arduino Enza e Franchini Angela Marinella, 1975. Controllo statistico di alcuni dati rilevati su terreni del piemonte. Istituto di Chimica Agraria di Torino.
Arduino Enza, Barberis Elisabetta, Ajmone Marsan Franco, Zanini Ermanno, and Franchini Marinella, 1986. Iron oxides and clay minerals within profiles as indicators of soil age in Northern Italy. Geoderma, 37: 45-55.
Asami Teruo, and Kubota Masatsugo, 1995. Background levels of soil berillium in several countries. Environmental Geochemistry and Health, 17: 32-38.
Atkinson R.J,, Posner A.M., and Quirk J.P., 1967. Adsorption of potential-determining ions at the ferric oxide-aqueous electrolite interface. Journal of Physical Chemistry, 71:550-558.
Atteia O., Th‚lin Ph., Pfeifer H.R., Dubois J.P., and Hunziker J.C., 1995. A search for the origin of cadmium in the soil of the Swiss Jura. Geoderma, 68: 149-172.
Bacon Jeffrey R., and Bain Derek C., 1995. Characterization of environmental water samples using strontium and lead stable isotope composition. Environmental Geochemistry and Health, 17: 39-49.
Baes C.F. Jr, and Mesmer R.E., 1976. “The Idrolysis of Cations”. John Wiley and Sons, New Tork. Citato in Bodec et al., 1988.
Baes C.F. III, 1982. Prediction of radionuclide Kd values from soil-plant concentration ratios. Trans. Amer. Nucl. Soc., 41: 53-54.
Baes C.F. III, Sharp R.D., Sjoreen A.L., and Shor R.W.A., 1984. A rewiew and alalysis of parameters for assesing transport of environmenthally released radionuclides through agricolture. Oak Ridge National Laboratory, TN, ORNL-5786.
Balistieri L. S. and Murray J. W., 1982. The adsortption of Cu, Pb, Zn, and Cd on goetite from major ion seaseawater. Geochimica et Cosmochimica Act, 46: 1253-1265.
Balistrieri L., Brewer P.G., and Murray J.W., 1981. Scavening residence time of trace metals and surface chemistry of sinking particles in the deep ocean. Deep-Sea Researches,28A: 101-121.
Balistrirei L.S., and Murray J.W., 1981. The surface chemistry of goetithe (alfaFeOOH) in maior ion seawater. American Journal of Science, 281: 788-806.
Barbieri F. Ed, 1983. “Structural model of Italy, scala 1:500000”, C.N.R., SENCA, Firenze.
Bar-Yosef B., S. fishman, and Talpaz H., 1970. A model for zinc movement to single roots in soils. Soil Science Society of America Journal, 44: 1272-1279.
Barber R.G., and Rowell D.L., 1972. Charge distribution and the cation exchange capacity of an iron-rich kaolinitic soil. Journal of Soil Science, 23: 135-146.
Beale A.M., Fasulo D.A., and Craigmill A.L., 1990. Effects of oral and parenteral selenium supplements on residues in meat, milk and eggs. Review of Environmental Contamination and Toxicology, 115: 125-150.
Beckwith R.S., 1959. Titration curves of soil organic matter. Nature, 184: 745-746.
Beckwith R.S., 1955. Metal complexes in soils. Australian Journal of Agricultural Researches, 6: 685-698.
Belzile N. and Tessier A., 1990. Interactions between arsenic and iron oxydroxides in lacustrine sediments. Geochimica et Cosmochimica Acta, 54: 103-109.
Belzile N., De Vitre R.R., and Tessier A., 1989. In situ collection of diagenetic iron and manganese oxydroxides from natural sediments. Nature, 340: 376-377.
Benjamim M.M., and Lekie J.O., 1981. A conceptual model for metal ligand-surface interactions during adsorption. Environmetal Science and Technology, 15: 1050-1057.
Benjamin M.M., and Leckie J.O., 1981. Multiple site adsorption of Cd, Cu, Zn,, and Pb on amorphous iron hydroxide. J. Coll. Inter. Sci., 79: 209-221.
Bertelsen B.O., Steinnes E., Solberg W., and Jingsen J., 1995. Heavy metal concentration in plants in relation to atmospheric heavy metals deposition. Journal of Environmenthal Quality, 24: 1018-1026.
Bewers J.M., Sundby B., and Yeats P.A., 1976. The distribution of trace metals in the wester north Atlantic of Nova Scotia. Geochimica et Cosmochimica Acta, 40: 687-696.
Bidwell A.M., and Dowdy R.H., 1987. Cadmium and zinc availability to corn following termination of sewage sludge applications. Journal of Environmental Quality, 16: 438-442.
Bifano C., and Mogoll J.L., 1995. Metallic contaminant profiles in sediment cores from Lake Valencia, Venezuela. Environmental Geochemistry and Health, 17: 113-118.
Birch Linda, Hanselmann Kurt W., and Bachofen Reinhard, 1995. Heavy metal conservation in lake Cadogno sediments: historical records of anthropogenic emission in a meromictic alpine lake. Water Research, 30: 679-687.
Birkeland P.W., 1983. “Soil and geomorfology”. Oxford University Press, New York.
Bittell J.F., and Miller R.J., 1974. Lead, cadmium and calcium selectivity coefficients on montmorillonite, illite and kaolinite. Journal of Environmenthal Quality, 12: 250-253.
Bloom P.R., and McBride M.B., 1979. Metal ion binding and exchange with hydrogen ions in acid-wasshed peat. Soil Science Society of America Journal, 43: 687-692.
Boast C.W., 1973. Modeling the movement of chemicals in soils by water. Soil Science, 115: 224-230.
Bodek I., Lyman W.J., Reehl F.W., and Rosenblatt D.H. Eds, 1988. “Environmental inorganic chemistry”, Pergamon Press, London.
Boile E.A., Scalter F.R., and Edmond J.M., 1977. The distribution of dissolved copper in the Pacific. Earth Planetary Science Letter, 37: 38-54.
Boile E.A., Scatler F., and Edmond J.M., 1976. On the marine geochemistry of cadmium. Nature, 263: 42-44.
Bolland M.D.A., Posner A.M., and Quirk J.P., 1976. Surface charge on kaolinites in aqueos suspension, Australan Journal of Soil Research, 14: 197-216.
Bolth G.H., Summer M. E., and Kamphorst A., 1963. A Study of the Equilibria Between Three Categories of Potassium in an Illitic Soil. Soil Science Proceedings.
Bolth G.H. Ed., 1979. “Soil Chemistry B. Physico-chemical models”, Elsevier, Amsterdam.
Bolth G.H., 1955. Ion adsorption by clays. Soil Science, 79: 267-276.
Bolton K.A., Sjoberg S., and Evans L.J., 1996. Proton binding and complexation constants for a soil humic acid using a quasi-particle model. Soil Science Society of America Journal, 60: 1064-1072.
Bonazzola G.C., Ropolo R., and Facchinelli A., 1993. Profiles and downward migration of 134Cs and 106Ru deposited on italian soils after the Chernobyl accident. Health Physycs, 64.
Borggaard O.K., 1974. Titrimetric determination of acidity and pK values of humic acid. Acta Chem. Scand., A28: 121-122.
Boswell F.C., 1975. Municipal sewage sludge and selected element applications to soil: effect on soil and fescue. Journal of Environmental Quality, 4: 267-273.
Bourg A.C.M. and Schindler P.W., 1979. Effect of ethylendiamminetetraacetic acid on the adsorption of copper(II) at amorphous silica. Inorganic Nuclear Chemical Letter, 15: 225-229.
Bourg A.C.M., and Schildler P.W., 1978. Ternary surfaces complexes. 1. Complex formation in the system silica-Cu(II)-ethylendiaminic. Chimia, 33: 166-168.
Bourg A.C.M., Joss S., and Schindler P.W., 1989. Ternary surface complexes. Complex formation in the system silica-Cu(II)-2,2 bipyridyl. Chimia, 33: 19-21.
Bowden J.W., Bolland D.A., Posner A.M., and Quirk J.P., 1973. Generalized anion and cation adsorption at oxide surface. Nature Physical Science, 245: 81-83.
Bowen H. J.. M., 1979. “Environmental Chemistry of the Elements”. Academic Press, London.
Bower C.A., Gardner W.R., and Goertzen J.O., 1957. Dinamic of cation exchange in soil columns. Soil Science Society of America Proceedings, 21:20-24.
Bowman R.S., and O'Connor G.A., 1982. Control of nickel and strontium sorption by free metal ion activity. Soil Science Society of America Journal, 46: 933-936.
Bowman R.S., Essington M.E., and O'Connor G.A., 1981. Soil sorption of nickel: influence of solution composition. Soil Science Society of American Journal, 45: 860-865.
Brandvold Lynn A., Popp Barbara R., and Swartz Sandra J., 1996. Lead content of plants and soils from three abandoned smelter areas in and near Socorro, New Mexico. Environmental Geochemistry and Health, 18:1-4.
Breuwsma A., and Liklema J., 1971. Interfacial electrochemistry of hematite (alfa-Fe2O3). Disc. Faraday Soc., 52: 324-333.
Brewer P.A., and Taylor M.P., 1997. The spatial distribution of heavy metal contaminated sediment across terraced floodplains. Catena, 30: 229-249.
Broadbent F.E., and Ott J.B., 1957. Soil organic matter complexes: 1. Factor affecting varius cations. Soil Science, 83: 419-427.
Brookins D. G., 1987.
“Eh-pH Diagrams for Geochemistry”. Springer-Verlag, Berlin.
Brouwer E., Baeyens B., Maes A., Cremers A., 1983. Cesium and Rubidium ion Equilibria in Illite Clay. Journal of Physical Chemistry, 7: 1213-1219.
Brown G., and Gastuche M.C., 1967. Mixed alluminium-magnesium hydroxides. II. Structure and structural chemistry of syntetic hydroxycarbonates and related mineral and compounds. Caly Minerals, 7: 198-201.
Brown-Anglin B., Brown-Armour A., and Lalor G.C., 1995. Heavy metal pollution in Jamaica: survey of cadmium, lead and zinc concentrations in the Kintyre and Hope Flat districts. Environmental Geochemistry and Health, 17: 51-56.
Browne M.A.E., and McMillan A.A., 1991. British geological survey thematic geology maps of quaternary deposits in Scotland. Quaternary Engineering Geology, Geological Society Engineering Geology Special Publication, 7: 511-518.
Browne Michael A.E., 1987. The physical geography and geology of the estuary and Firth of Forth, Scotland. Proceedings of the Royal Society of Edinburgh, 93b: 235-244.
Bruland K.W., Knauer G.A., and Martin J.H., 1978. Cadmium in northeast Pacific waters. Limnol. Oceanogr., 23: 618-625.
Bruland K.W., Knauer G.A., and Martin J.H., 1978. Zinc in northeaster Pacific waters. Nature, 271: 741-743.
Bruno E., and Facchinelli A., 1972. Al, Si Configuration in Lead Feldpar. Zirfricht Kristallografica, 136: 296-300.
Buaur W.H. e Onishi H., 1972. “Arsenic”. In: Whedepohl K.H. Ed., 1972. “Handbook of Geochemistry”, Springher-Verlag, Berlin.
Buchet Jean P., Lauwerys Robert R., and Yager Janice W., 1995. Lung retention and bioavailability of arsenic after single intratracheal administration of sodium arsenite, sodium arsenate, fly ash and copper smelter dust in hamster. Environmental Geochemistry and Health, 17: 182-188.
Busetti G., 1983. “Esercitazioni pratiche di fisica”, Libreria Editrice Universitaria Levrotto e Bella, Torino.
Callahan M., Slimak M., Gabel N., May I., Fowler
C., Freed J., Jennings P., Durfee R., Whitmore F., Maestri B., Mabey W., Holt
B., and Gould C, 1979. “Water-related environmental fate of 129 priority
pollutants. Volume I: introduction and technical background, metals and
inorganics, pesticides and PCBs”. EPA-440/4-79-029a, EPA contracts 68-01-3852
and 68-01-3867, Office of Water Planning and Standards, U.S. Environmenntal
Protection Agency, Wascington D.C. Citato in Bodec I. et al., 1988.
Cameron B.I., and Stephenson D, 1985. “The midland valley III edition”. British Geological Survey, London.
Candelaria L.M., Chang A.C., and Amrhein C., 1995. Measuring cadmium ion activities in sludge-amended soils. Soil Science, 159: 162-175.
Cao H.F., Chang A.C., and Page A.L., 1984. Heavy metaL contents of sludge-treated soils as determined by three extraction procedure. Journal of Environmental Quality, 13: 632-634.
Carignan R., and Nriagu J.O., 1985. Trace metal deposition and mobility in the sediments of two lakes near Sudbury, Ontario. Geochimica et Cosmochimica Acta, 49: 1753-1764.
Carignan R., Rapin F., and Tessier A., 1985. Sediment porewater sampling for metal analysis: a comparision of tecniques. Geochimica et Cosmochimica Acta, 49: 493-2497.
Carpenter R.H., Robinson G.D., and Hayes W.B., 1978. Partitioning of Mn, F, Cu, Zn, Pb, Co, and Ni in black coatings on stream boulders in the vicinity of the Magrunder mine, Lincoln Co., Georgia. Journal of Geochemical Exploration, 10: 75-89.
Cashen G.H., 1966. Electric charges of clays. Journal of Soil Science, 17: 303-316.
Castiglioni G.B., 1986. “Geomorfologia, II edizione”. UTET, Torino.
Catts J.G., and Langmuir D., 1986. Adsorption of Cu, Pb, and Zn by delta-MnO2: applicability of the site binding-surface complexation model. Applied Geochemistry, 1: 255-264.
Chang A.C., Warneke J.E., Page A.L., and Lund L.J., 1984. Accumulation of heavy metals in sewage sludge-treated soils. Journal of Environmental Quality, 13: 87-91.
Chao T. T., 1972. Selective dissolution of manganese oxides from soils and sediments with acidified hydroxilamine hydrochloride. Soil Science Society of America Proceedings, 36: 764-768.
Cihacek L.J., and Bremner J.M., 1979. A simplified ethylen glycol monoethyl ether procedure for assesment of soil srface area. Soil Science Society of America Proceedings, 23: 821-822.
Coleman N.T., Weed S.B., and McCracken R.J., 1959. Cation exchange capacity and exchangeable cations in piedmont soils of North Caroline. Soil Science Society of America Proceedings, 23: 146-149.
Coughtrey P.J.,
Jackson D.J., Thorne M.C., 1985. “Radionuclide Distribution and Transport
in Terrestrial and Acquatic Ecosistems, a Compendium of Data”. 6 Vol. Commision
of the European Communities Directorate - General Information Market and Innovation, Luxemburg.
Cowan C.E., Zacharia J.M., and Resch C.T., 1991. Cadmium adsorption on iron oxides in the presence of alkaline-earth elements. Environmetal Science and Technology, 25: 437-446.
Cremers A., Elsen A., De Preter P., and Maes A., 1988. Quantitative Analysis of Radiocesium Retention in Soils. Nature, 355: 247-249.
Cushman J.H., 1979. An analitytical solution to solute transport near root surfaces for low initial concentration: 1. Equation development. Soil Science Society of America Journal, 43: 1087-1090.
D’Amico C., Innocenti F., Sassi F.P., 1987. “Magmatismo e metamorfismo”, UTET, Torino.
Davies B.E., and Lewin J., 1974. Chronosequences in alluvial soils with special reference to historic lead pollution in Cardiganshire, Wales. Environmet Pollution, 6: 49-57.
Davies B.E., 1992. “Lead”. In: “Alloway B.J. Ed., 1992. “Heavy Metals in Soils”, Blackie Academic and Profesional, London.
Davis J.A., 1984. Complexation of trace metals by adsorbed natural organic matter. Geochimica et Cosmochimica Acta, 48: 679-691.
Davis J.A., 1982. Adsorption of natural dissolved organic matter at the oxide/water interface. Geochimica et Cosmochimica Acta, 46: 2381-2393.
Davis J.A. and Leckie J.O., 1978. Surface ionization and complexation at the oxide/water interface. 2. Surface properties of amorphous iron oxydroxide and adsorption of metals ions. Journal of Colloid Interface Science, 67: 90-105.
Davis J.A., and Leckie J.O., 1980. Surface ionization and complexation at the oxide/water interface. 3. Adsorption of anions, Journal of Colloid Interface Science, 74: 32- .
Davis J.A., and Leckie J.O., 1978. Effect of adsorbed complexing ligands on trace metals uptake by hydrous oxides. Environmetal Science and Tecnology, 12:1309-1315.
Davis J.A., Fuller C.C., and Cook A.D., 1987. A model for trace metal sorption processes at the calcite surface: adsorption of Cd2+ and subsequent solid solution formation. Geochimica et Cosmochimica Acta, 51: 1477-1490.
Davis J.A., James R.O., and Leckie J.O., 1978. Surface ionization and complexation at the oxide/water interface. 1. Computation of electrical double layer properties in simple electrolytes. Journal of Colloid Interface Science, 63: 480-499.
Davison W., 1993. Iron and manganese in lakes. Earth-Science Review, 34: 119-163.
Davison W., Grime G.W., Morgan J.A., and Clarke K., 1991. Distribution of dissolved iron in sediment pore waters at submillimetre resolution. Nature, 352: 323-325.
Day G.M., Hart B.T., McKelvie I.D., and Beckett R., 1994. Adsorption of natural organic matter onto goethite. Colloids Surf. A: Physicochem. Engineer. Aspects, 89: 1-13.
De Leenheer L., and De Boodt M., 1951. Characterization of the belgian sea-polder soils by the clay fraction and cation-exchange capacity. Journal of Soil Science, 2: 183-187.
Delgado Garcia R.A., Herruzo Garcia F, Maroto Rodriguez J.M., and Vereda C., 1996. Influence of soil carbonates in lead fixation. Journal of Environmental Science and Health, A31: 2099-2109.
Delgado M., Bigeriego M., and Guardiola E., 1993. Uptake of Zn, Cr, and Cd by water hyacinths, Water Research, 27: 269-272.
DiToro D.M., Mahony J.D., Kirchgraber P.R., O'Birne A.L., Pascuale L.R., and Piccirilli D.C., 1986. Effects of non reversibility, particle concentration, and ionic strength on heavy metal sorption. Environmenthal Science and Technology, 20:55- .
Dolcater D.L., Jackson M.L., and Syers J.K., 1972. Cation exchange selectivity in mica and vermiculite. American Mineralogist, 57: 1823-1831.
Domingo Jose L., 1989. Cobalt in the environment and its toxicological implication. Review of Environmental Contamination and Toxicology, 108: 105-131.
Doner H.E., 1978. Chloride as factor in mobilities of Ni(II), Cu(II), and Cd(II) in soil. Soil Science Society of America Journal, 42: 882-885.
Dormaar J.F., 1964. Fractionation of humus of some chernozemic soils of Suthern Alberta. Canadian Journal Soil Science, 44: 232-236.
Dowdy R.H., and Larson W.E., 1975. Metals uptake by barley seedlings grown on soils amended with sewage sludge. Journal of Environmental Quality, 4: 229-233.
Dreicer M., Haconson T.E., White G.C., and Whicker F.W., 1984. Rainsplash as a mechanism for soil contamination of plant surfaces. Health Physics, 46: 177-178.
Drew M.C., Nye P.H., and Vaidyanathan L.C., 1969. The supply of nutrient ions by diffusion to plant roots in soils. 1. Adsorption of potassium by cylindrical roots of onions and leek. Plant and Soil, 30: 252-270.
Edginton D.N., and Robbins J.A., 1976. Records of lead deposition in Lake Michigan since 1800. Environmental Scienc and Technology, 10: 226-274.
Egozy Y., 1980. Adsorption of cadmium and cobalt on montmorillonite as a function of solution composition. Clays and Clay Minerals, 28: 311-317.
Eisenman G., 1962. Cation selective glass electrodes and theyr mode of operation. Biophysical Journal supplement part II, 2: 259-323.
Eklund Mats, 1995. Cadmium and lead deposition around a Swedish battery plant as recorded in Oak tree rings. Journal of Environmenthal Quality, 24: 126-131.
Elder J.F., Perry S.K., and Brady F.P., 1975. Application of energy-dispersive x-ray fluorescence trace metal analysis of natural waters. Environmental Science and Technology, 9: 1039-1042.
Emmerich W.E., Lund L.J., Page A.L., and Chang A.C., 1982. Movement of heavy metals in sewage sludge treated soils. Journal of Environmental Quality, 11: 174-178.
Epstein E., and Hagen C.E., 1952. A kinetic study of the absorption of alcali cations by barely roots. Plant Physiology, 40: 620-624.
Esser K.B., Bockheim J.G., and Helmke P.A., 1991. Trace elements distribution in soils formed in the indiana dunes, U.S.A.. Soil Science, 152: 340-350.
Ewans D.W., James J. A., and Clark R.A. III, 1983. Reversible Ion-Exchange Fixation of Cesium-137 Leading to Mobilization From Reservois Sediments. Geochimica et Cosmochimica Acta, 47: 1041-1049.
Evans L.J., 1989. Chemistry of metal retention by soils. Environmental Science and Technology, 23: 1046-1056.
Farmer Jong G., and Lorna Eades J., 1996. Stable lead isotope record of lead pollution in loch Lommond sediments since 1630 A.D.. Environmental Science and Technology. In stampa.
Farrah H., and Pickering W.C., 1976. The sorption of copper species by clays. I. Kaolinite. Aust. J. Chem., 29: 1167-1176.
Farrar D.M., and Coleman J.D., 1967. The correlation of surface area with other properties of nineteen British clay soils. Journal of Soil Science, 18: 118-124.
Faure G., 1992. “Principles and application of inorganic geochemistry”. Maxwell Mcmillan International Editions.
Feddes R.A., Kabat P., van Bakel P.J.T., Bronswijk J.J.B., and Halbertsma J., 1988. Modellling soil water dynamics in the unsaturated zone- state of the art. Journal of Hydrology, 100: 69-111.
Felbeck J.T. Jr, 1971. Structural hypotheses of soil humic acids. Soil Science, 111: 42-48.
Fendorf Scott E., Li Guangchao, and Gunter Mickey E., 1996. Micromorphologies and stabilities of chromium (III) surface precipitates elucidated by scanning force microscopy. Soil Science Society of America Journal, 60: 99-106.
Ferris A.P., and Jepson W.B., 1975. The exchange capacities of kaolinite and the preparation of homoionic clays. Journal of Colloid Interface Science, 51: 245-259.
Flyhammar Peter, 1995. Analysis of the cadmium flux in Sweden with special emphasys on landfill leachate. Journal of Environmenthal Quality, 24: 612-621.
Forbes E., Posner A.M., and Quirk J.P., 1974. The specific adsorption of inorganic Hg(II) species and Co(II) complexes ion on goetite. Journal of Colloid Interface Science, 49: 403-409.
Forbes E.A., Posner A.M., and Quirk J.P., 1976. The specific adsorption of divalent Cd, Co, Cu, Pb, and Zn on goethite. Journal of Soil Science, 27: 154-166.
Fortin D., Leppard G.G., and Tessier A., 1993. Characteristics of lacustrine diagenetic iron oxyhydroxides. Geochimica et Cosmochimica Acta, 57: 4391-4404.
Francani V., 1988. “Geologia applicata 4. Idrogeologia generale”. CLUP, Milano.
Francis C. W., and Brincley F.S., 1976.
Prefertial Adsorption of 137Cs
in Contaminated Freshwater Sediments. Nature, 260: 511-513.
Francoise C. Richard, and Bourg Alain C.M., 1991. Aqueous geochemistri of cromium: a rewiew. Water Research, 25: 807-816.
Frega G., 1987. “Aspetti idrologici e idraulici della pianificazione nei bacini idrografici”. Editoriale BIOS, Cosenza.
Fu G., Allen H.E., and Cowan C.E., 1991. Adsorption of cadmium and copper by manganese oxides. Soil Science, 152: 72-81.
Gadde R.R. and Laitinen H. A., 1974. Studies of havy metal adsorption by hydrous iron and manganese oxides. Analitical Chemistry, 42: 2022-2026.
Garcia-Miragaya and Page A. L., 1977. Influence of exchangeable cation on the sorption of trace amounts of cadmium by montmorillonite. Soil Science Society of America Journal, 41: 718-721.
Garcia-Miragaya J., and Page A.L., 1976. Influence of ionic strength and inorganic complex formation on the sorption of trace amounts of Cd by montmorillonite. Soil Scince Society American Jounal, 40: 658-663.
Garcia-Miragaya J., and Page A.L., 1977. Influence of exchangeable cation on the sorption of trace amounts of cadmium by montmorillonite. Soil Science Society of America Journal, 41: 718-721.
Garrels R.M., and Thompson M.E., 1962. A chemical model for seawater. American Journal of Sciene, 260: 57-66.
Gast R.G., 1972. Alkali metal cation exchange on chambers montmorillonite. Soil Science Society of America Proceedings, 36: 14-19.
Gessa C. e Testini C., 1989a. “Adsorbimento Anionico”. In: Sequi P. Ed., 1989. “Chimica del suolo”, Patròn Editore, Bologna.
Gessa C. e Testini C., 1989b. “Lo scambio cationico”. In: Sequi P. Ed., 1989. “Chimica del suolo”, Patròn Editore, Bologna.
Gibbs R.J., 1977.
Transport Phase of Transition Metals in the Amazon and Yukon Rivers. Bulletin
of Geological Society of America, 88: 829-843.
Goldberg E.D., 1965. “Minor Elements in Seawater”. In: Riley J.P. and Skiroow G., 1965. “Chemical Oceanography”, Academic Press, London. Citato in Wedepohl K.H. Ed., 1978.
Goldberg S., 1986. Chemical modeling of arsenate adsorption on alluminium and iron oxide minerals. Soil Science Society of America Journal, 50: 1154-1157.
Goldberg S., and Glaubig R.A., 1988. Anion adsorption on a calcareus, montmorillonitic soil- Arsenic. Soil Science Society of America Journal, 52: 1157-1297.
Gongmin Fu, Allen Herbert E., and Cowan Christina E., 1991. Adsorption of cadmium and copper by manganese oxide. Soil Science, 152: 72-81.
Graham R.C., Diallo M.M., and Lund L.J., 1990. Soils and mineral weathering on phyllite colluvium and serpentinite in Northwestern California. Soil Science Society of America Journal, 54: 1682-1690.
Greenland D. J., 1975. Charge characteristics of some kaolinite-iron hydroxides complexes. Clay Mineralogy, 10: 407-416.
Greenland D.J., 1965. Interaction between clays and organic compounds in soils. Part II. Adsorption of soil organic compounds and its affects on soil properties. Soils Fert., 28: 521-532.
Grutter A., Von Gunten R., and Rossler E., 1986. Sorption, Desorption and Isotope Exchange of Cesium (10^-9-10^-3 M) on Chlorite. Clays and Clay Mirerals, 34: 677-680.
Gu B., Schmitt J., Chen Z., Liang L., and McCarthy J.F., 1994. Adsorption and desorption of natural organic matter on iron oxide: mechanism and models. Environmental Sciece and Technology, 28: 38-46.
Gulmini M., Zelano V., Daniele P.G., Prenesti E., Ostacoli G., 1996. Acid-base properties of a river sediment: applicability of potentiometric titrations. Analytica Chimica Acta, 329: 33-39.
Hammond P.B., and Dietrich K.N., 1990. Lead exposure in early life: health consequences. Review of Environmental Contamination and Toxicology, 115: 191;124.
Hargrove W.L., and Thomas G.W., 1981. Extraction of alluminium from alluminium-organic matter complexes. Soil Science Society of America Journal, 45: 151-153.
Harter R.D., and Baker D.E., 1977. Applications and misapplications of the Langmuir equation to soil adsorption phenomena. Soil Science Society of America Journal, 41: 1077-1080.
Harter Robert D., 1993. Effect of soil pH on adsorption of lead, copper, zinc, and nickel. Soil Science Society of America Journal, 47: 47-51.
Hawley J.K., 1985. Assesment of health risk from exposure to contaminated soil. Risk Analysis, 8: 289-302.
Heckman J.R., Angle J.S., and Chaney R.L., 1987. Residual effects of sewage sludge on soybean: I. Accumulation of heavy metals. Journal of Environmental Quality, 16: 113-117.
Heilman M.D., Carter D.L., and Gonzalez C.L., 1965. The ethylene glycol monoethyl ether (EGME) tecnique for determining soil surface area. Soil Science, 100: 409-413.
Henrichs H., Schoultz-Dobrick B., and Wedepohl K.H., 1980. Terrestrial geochemistry of Cd, Bi, Tl, Pb, Zn, and Rb. Geochimica et Cosmochimica Acta. 44: .
Hendricson L.L., and Corey R.B., 1981. Effect of equilibrium metal concentrations on apparent selectivity coefficient of soil complexes. Soil Science, 131: 163-171
Hickey M.G. and Kittric J.A., 1984. Chemical partitioning of cadmium, copper, nickel, and zinc in soils and sediments containing high levels of heavvy metals. Journal of Environmenthal Quality, 13: 372-376.
Higgins A.J., 1984. Land application of sewage sludge with regard to cropping system and pollution potential. Journal of Environmental Quality, 13: 441-448.
Hill D.E., and Sawhney B.L., 1969. Electron Micropobe Analysis of Thin Section of Soil to Observe Loci of Cation Exchange. Soil Science Society Proceedings, 33: 531-534.
Hilts Steven R., 1996. A co-operative approach to risk managment in an active lead/zinc smelter community. Environmental Geochemistry and Health. 18: 17-24.
Hingston F.J., Posner A.M., and Quirk J.P., 1971. Competitive adsorption of negatively charged ligands on oxide surfaces. Discuss. Faraday Soc., 52: 334-342.
Hodgon J.F., 1960. Cobalt reaction with montmorillonite. Soil Science American Journal Proceedings, 24: 165-168.
Hodgson J.F., 1963. "Chemistry of the micronutrient elements in soils". Advances in Agronomy, 15: 119-159.
Holm P.E., Andersen B.H., and Christensen T.H., 1996. Cadmium solubility in aerobic soils. Soil Science Society of America Journal, 60: 775-780.
Holm P.E., Christenses T.H., Tjell J.C., and McGrat S.P., 1995. Speciation of cadmium and zinc with application to soil solution. Journal of Environmenthal Quality, 24: 183-190.
Huang Tien-Shang, Lu Fung-Jou, and Tsai Chan-Wu, 1995. Tissue distribution of absorbed humic acids. Environmental Geochemistry and Health, 17: 1-4.
Huerta-Diaz M.A., Tessier A., and Carignan R., 1996. Geochemistry of trace metals associated with reduced sulfur in freshwater sediment. Geochimica et Cosmochimica Acta. In stampa.
Hunsaker V.E., and Pratt P.F., 1970. The formation of mixed magnesium-alluminium hydroxides in soil materials. Soil Science Society of America Proceedings, 34: 813-816.
Ibekwe A.M., Angle J.S., Chaney R.L., and van Berkum P., 1995. Sewage sludge and heavy metal effects on nodulation and nitrogen fixation of legumens. Journal of Environmenthal Quality, 24: 1199-1204.
Inskeep W.P., and Baham J., 1983. Adsorption of Cd(II) and Cu(II) by montmorillonite at low surface coverage. Soil Science Society American Journal, 47: 660-665.
Irvig H. and Williams R. J. P., 1948. Order of stability of metal complexes. Nature, 162: 746-747.
Iyengard S.S., Martens D.C., and Miller W.P., 1981. Distribution and plant availability of soil zinc fractions. Soil Science Society of American Journal, 45: .
Jackson M. L., 1963. Interlayering of expansibile layer silicates in soils by chemical wathering. In: “Eleventh National Conference on Clays and Clay Minerals”. Earl Ingerson Ed., Pergamon Press.
Jackson D.R., and Watson A.P., 1977. Disruption of nutrient pools and transport of heavy metals in a forest watershed near a lead smelter. Journal of Environmental Quality, 6: 331- .
James Bruce R., 1994. Hexavalent chromium solubility and reduction in alcaline soils enriched with chromite ore processing residue, Journal of Environmental Quality, 23: 227-233.
James R.O., and Healey T.W., 1972. Adsorption of hydrolizable metal ions at the oxide-water interface. Journal of Coll. Inter. Sci., 40: 42-81.
James R.O., Stiglich R.J. and Healy T.W., 1975. Analysis of model of adsorption of metal ion at oxide/water interfaces. Farad. Disc. Chem. Soc., 59: 142-156.
Jie X.L., Liu F., Zhou D.H., Xu F.L., Li X.Y., and Wang D.F., 1995. Transformation of coordiante forms of phosphate adsorbed on goethite surface under condition of varyng pH. Pedosphere, 5: 229-235.
Jin X.C., Bailey G.W., Yu Y.S., and Lynch A.T., 1996. Kinetics of single and multiple metal ion sorption processes on humic substances. Soil Science. In stampa.
Jin X.C., Shu N.N., Wu S.D., and Wang C.C., 1987. Study on the rate of adsorption and desorption of heavy metals, cadmium, and mercury by sediment suspension from Xiangjing River. Chinese Environmental Sciene, 7: 21-26.
Johnson B.B., 1990. Effect of pH, temperature, and concentration on the adsorption of cadmium on goethite. Environmental Science and Technology, 24: 112- .
Jones K.C., Peterson P.J., and Davies B.E., 1984. Extraction of silver from soils and its determination by atomic absorption spectrometry. Geoderma, 33: 157-168.
Jones R.L., Hinesly T.D., Ziegler E.L., and Tyler J.J.. 1975. Cadmium and zinc contents of corn leaf and grain produced by sludge-amended soil. Journal Environmental Quality, 4: 509-514.
Kan S.U., 1969. Some chemical characteristics of humic acids extracted from a solonetz-solod-black soil sequence at Vegreville, Alberta. Canadian Journal of Soil Sciemce, 49: 168-170.
Karathanasis A.D., and Wells K.L., 1989. A comparision of mineral weathering trends between two managment systems on a catena of loess-derived soils. Soil Science Society of America Journal, 53: 582-588.
Kawaguchi K., and Kyuma K., 1959. On the complex formation between soil humus and polyvalent cations. Soil Plant Food, 5: 54-63.
Keller C., and Vedy J.C., 1994. Distribution of copper and cadmium fractions in two forest soils. Journal of Environmenthal Quality, 23: 987-999.
Kelling K.A., Keeney D.R., Walsh L.M., and Ryan J.A., 1977. A field study of the agricultural use of sewage sludge: III. Effect on uptake and extractability of sludge-borne metals. Journal of Environmental Quality, 6: 352-358.
Kelly J., and Thorton I., 1996. Urban geochemistry: a study of the influence of antropogenic activity on the heavy metal content of soils in traditionally industrial and non-industrial areas of Britain. Applied Geochemistry, 11: 363-370.
Kerndorff H., and Schnitzer M., 1980. Sorption of metals on humic acid. Geochimica et Cosmochimica Acta, 44: 1701-1708.
Khan S.U., 1969. Interaction between the humic acid fraction of soils and certainn metallic cations. Soil Science Society of America Journal, 33: 851-854.
Khanna S.S., and Stevenson F.J., 1962. Metallo-organic complexes in soil: 1. Potentiometric titration of same soil organic matter isolates in the presence of transition metals. Soil Science, 93: 298-305.
Kinniburg D.G., Jackson M.L., and Syers J.K., 1976. Adsorption of alcaline earth, transition, and hevy metal cations by hydrous oxide gels of iron and alluminium. Soil Science Society of America Journal, 40: 796-799.
Kinniburg D.G., Syers J.K., and Jackson M.L., 1975. Specific adsorption of trace amounts of calcium and strontium by hydrous oxides of iron and alluminium. Soil Science Society of America Procedings, 39: 464-470.
Kinniburg D.G., Syers J.K., and Jackson M.L., 1975. Specific adsorption of trace amounts of strontium and calcium by hydrous oxides of iron and alluminium. Soil Science Society of Americ Proceedings, 39: 464-470.
Kirkham M.B., 1975. Trace elements in corn grown on long-term sludge disposal site. Journal Environmental Quality, 9: 765-768.
Kittrick J.A., 1966. Forces involved in ion fixation by vermiculite. Soil Sience Society of America Proceedings, 30: 801-803.
Klobe W.D., and Gast
R.G., 1970. Condition Affecting Cesium Fixation and Sodium Entrapment in
Hydrobiotite and Vermiculite. Soil Science
Society Proceedings, 34: 746-750.
Koide M., soutar A., and Goldberg E.D., 1972. Marine geochronology with 210-Pb". Earth Planet Science Letters, 14: 2402-2413.
Kolthoff I.M., and Overholser L.G., 1939. Studies on aging and coprecipitation. XXVIII. Adsorption of divalent ions on and coprecipitation with ortho ferric hydroxide in ammoniacal medium. Journal of Physical Chemistry, 43: 767-780.
Komarneni S., 1978. Cesium sorption and desorption behavior of kaolinites. Soil Science Society American Journal, 42: 531-532.
Kong In-Chul, Gabriel Bitton, Koopman Ben, and Jung Keum-Hee, 1995. Heavy metal toxicity testing in environmental samples. Review of Environmental Contamination and Toxicology, 142: 119-147.
Konshin O. V., 1982. Appplicability of the convection-diffusion mechanism foe modelling migration of 137-Cs and 90Sr in the soil. Helth Physics, 63: 291-300.
Korte N.E., Skopp J., Fuller W.H., Niebla E.E., and Alesii B.A., 1976. Trace element movement in soils: influence of soil Physical and chemical properties. Soil Science, 122: 350-359.
Korte N.E., Skopp J., Niebla E.E., and Fuller W.H., 1975. A baseline study on trace metal elution from diverse soil types. Water, Air, Soil Pollution, 5: 149-156.
Koyama Macoto, 1995. Adsorption of humic acid on Ca-Montmorillonite. Soil Science and Plant Nutrition, 41: 215-223.
Kuo S., Heilman P.E., and Baker A.S., 1983. Distribution and forms of copper, zinc, cadmium, iron, and manganese in soils near a copper smelter. Soil Science, 135: 101-109.
Kupcic V., Ahrens L.H., Earlank A.J.,, Wedepohl K.H., 1978. “Arsenic”. In: Wedepohl K.H. Ed., “Handbook of geochemistry, vol. II-5”. Springer-Verlag, Berlin.
Lantzy R.J., and Mckenzie F.T., 1979. Atmosferic trace metals: global cycles and assesment of man’s impact. Geochimica et Cosmochimica Acta, 43: 511-525.
Lasaga A.C., 1980. The kinetic treatment of geochemical cycles. Geochimica et Cosmochimica Acta, 44: 815-828.
Le Riche H. H. and Weier A. H., 1963. A method of studing trace elements in soil fractions. Journal of Soil Science, 14: 225-234.
Le Roux J., Rich C.I., 1969. Ion selectivity of micas as influenced by degree of potassium depletation. Soil Science Society Proceedings, 33: 684-689.
Le Roux J., Rich C.I., and Ribbe P. H., 1970. Ion selectivity by weathered micas as determened by electron microprobe analisis. Clays and Clay Mineral, 18: 333-338.
Lee R., Bache B.W., Wilson M.J., and Sharp G.S., 1983. Alluminium release in relation to the determination of cation exchange capacity of same podzolized New Zeland soils. Journal of Soil Science, 36: 239-253.
Lee Robert C., Fricke James R., Wright William E., and Haerer Walt, 1995. Development of a probabilistic blood lead prediction model. Environmental Geochemistry and Health, 17: 169-181.
Leenaers H., Scouten C.J., Rang M.C., 1988. Variability of the metal content of flood deposits. Environ. Geol. Water Sci., 11: 95-106.
Leinweber P., Reuter G., and Brozio K., 1993. Cation exchange capacities of organo-mineral particle-size fractions from long-term experiment. Journal of Soil Science, 44: 111-119.
Lerman A., and Mackenzie F.T., 1975. Modeling of geochemical cycles: phosphorus as an example. Geological Society of America, 142: 205-217.
Le Roux J., Rich C.I., 1969. Ion Selectivity of Micas as influenced by degree of potassium depletetion. Soil Science Society Proceedings, 33: 684-689.
Levi-Minzi R. e Riffaldi, 1989. “Il controllo degli inquinanti in agriccoltura”. In: Sequi P., 1989. “Chimica del suolo”, Patron Editore, Bologna.
Lewandowski Thomas A., and Forlsund Barbara L., 1994. Comparision of IEUBK model predictions and actual blood values at a former battery recycling site. Environmental Geochemistry and Health, 16: 217-222.
Lichtner P. C., 1988. “Modeling reactive flow and transport in natural systems”. In: Marini L. e Ottonello G. Eds, 1996. “Proceedings of the Rome Seminar on environmental geochemistry”, Castelnuovo di Porto, May 22-26 1997.
Li Yuan-Hui, 1982. Interelement relationship in abyssal Pacific ferromanganese nodules and associated pelagic sediments. Geochimica et Cosmochimica Acta, 46: 1053-1060.
Lilia Albert A., and Badillo Francisco, 1991. Environmental lead in Mexico. Review of Environmental Contamination and Toxicology, 115: 91-124.
Lim C.H., Jackson M.L., Koons R.D, and Helmke P.A., 1980. Kaolins: sources of differences in cation-exchange capacities and cesium retention. Clays and Clay Minerals, 28: 223-229.
Lin Chiu-Yue, 1993. Effect of heavy metals on acidogenesis in anaerobic digestion. Water Research, 1: 147-152.
Liu F., Jie X.L., Zhou D.H., Xu F.L., Li X.Y., He J.Z., and Wang D.F., 1995. Influence of pH on chemical forms of phosphate adsorbed on goethite surface. Pedosphere, 5: 157-162.
Loganatha P., and Burau R.G., 1973. Sorption of heavy metals by hydrous manganese oxide. Geochimica et Cosmochimica Acta, 37: 1277-1293.
Lopez-Hernandez D., Siegert G., and Rodriguez J.V., 1986. Competitive adsorption of phosphate with malanate and oxalate by tropical soils. Soil Science Society of America Journal, 50: 1460-1462.
Louma S.N., 1986. A comparision of two methods for determining copper partitioning in oxidized sediment. Mar. Chem., 20: 45-49.
Louma S.N. and Davis J.A., 1983. Requirements for medelling trace metals partitioning in oxidized estuarine sediments. Mar. Chem., 12: 159-181.
Lowe L.E., 1969. Distribution and properties of organic fractions in selected Alberta soils. Canadian Journaal of Soil Science, 49: 129-141.
Lumsdon David G., Evans Leslie J., and Bolton Kim A., 1995. The influence of pH and chloride on the retention of cadmium, lead, mercury, and zinc by soils. Journal of Soil Contamination, 4: 1-14.
Lund L.J., Page A.L., and Nelson C.O., 1976. Movement of heavy metals below sewage disposal ponds. Journal of Environmental Quality, 1976: 330-334.
Ma Q.Y., and Lindsay W.L., 1995. Estimation of Cd2+ and Ni2+ activities in soils by chelation. Geoderma, 68: 123-133.
Mackay H.A.C., 1938. Kinetics of exchange reactions. Nature, 142: 997-998.
Mackenzie F.T., Lantzy R.J., Paterson V., 1979. Global trace metal cycles and predictions. Journal of International Association of Mathematical Geology, 11: 99-142.
Madoni Paolo, Davoli Donatella, Gorbi Gessica, and Vescovi Luciano, 1996. Toxic effect of heavvy metals on the activated sludge protozoa community. Water Research, 30: 135-141.
Mahaney William C., and Hancock R.G.V., 1996. Geochemical evidence of pollutants from coal-fired generating station in late Pleistocene palaeosols in the Dalijia Shan, northwestern China, Environmental Geochemistry and Health, 18: 35-40.
Maier Kurt J., and Knight Allen W., 1994. Ecotoxicology of Selenium in freshwater systems. Review of Environmental Contamination and Toxicology, 134: 31-48.
Maione U., 1995. “Le piene fluviali”, La Goliardica Pavese, Pavia.
Mann S.S., and Ritchie G.S.P., 1993. The influence of pH on the forms of cadmium in four west australian soils. Australian Journal of Soil Research, 31: 255-270.
Manning B.A., and Goldberg S., 1966. Modeling competitive adsorption of arsenate with phosphate and molybdate on oxide minerals. Soil Science Society of America Journal, 60: 121-131.
Manning B.A., and Goldberg S., 1966. Modeling arsenate competitive adsorption on kaolinite, montmorillonite and illite. Clays and Clay Minerals, 44: 609-623.
Mantoura R.F.C., and Riley J.P., 1975. The use of the gel filtration in the study of metal binding by humic acids and related compounds. Analitical Chimica Acta, 78: 193-200.
Mantoura R.F.C., Dickson A., and Riley J.P., 1978. The complexation of metals with humic materials in natural waters. Estuar. Coast. Mar. Sci., 6: 387-408.
Manunza Bruno, Deiana Salvatore, Maddau Vittoria, Gessa Carlo, and Seeber Renato, 1995. Stability constant of metal-humate complexes: titration data analyzed by bimodal gaussian distribution. Soil Science Society of America Journal, 59: 1570-1574.
Marsan Ajmone Franco, Barberis Elisabetta, and Arduino Enza, 1988. A soil chronosequence in Northwestern Italy: morphological, physical and chemical characteristics. Geoderma, 42: 51-64.
Maskall John, Whitehead Keith, and Thornton Ian, 1995. Heavy metal migration in soils and rocks at historical smelting sites. Environmental Geochemistry and Health, 17: 127-138.
Mastrangelo F., Natale P., e Zucchetti S., 1983. Quadro giacimentologico e metallogenico delle alpi occidentali italiane. Associazione Mineraria Subalpina, marzo-giugno 1983, n° 1-2.
Mathur S.P., and Levesque M.P., 1983. The effects of using copper for mitigating histosol subsidence on: 2. The distribution of copper, manganese, zinc, and iron in an organic soil, mineral sublayers, and their mixtures in the context of setting a threshold of phytotoxic soil-copper. Soil Science, 135: 166-176.
Matsuda K., and Ito S., 1970. Adsorption strength of zinc for soil humus; III. Relationship between stability constants of zinc-humic and -fulvic acid complexes and degree of humification. Soil Science and Plant Nutrition, 16: 1-10.
Mattigot S.V., Pratt P.V., and Schalsha E.B., 1985. Trace metal speciation in soil profile irrigated with waste waters. Water Science and Tecnology, 17: 133-142.
Mattson S., 1931. The laws of soil colloidal behavior: V. Ion adsorption and exchange. Soil Science, 31: 311-331.
Mayer L.M., 1994. Relationship between mineral surfaces and organic carbon concentration in soils and sediments. Chem. Geol., 114: 347-363.
Mayer L.M., 1994. Surface area control of organic carbon concentrations in soils and sediments, Geochimica et Cosmochimica Acta, 58: 1271-1284.
Mc Grath P., and Cegarra J., 1992. Chemical extractability of heavy metals during and after long-term applications of sewage sludge to soil. Journal of Soil Science, 43: 313-321.
Mc Intire W.L., 1963. Trace element partition coefficents- a rewiew of theory and application to geology. Geochimica et Cosmochimica Acta, 27: 1209-1264.
McAleese D.M., 1958. Studies on the basaltic soils of northern ireland VI. Cation-exchange capacities and mineralogy of the fine-sand separates. Journal of Soil Science, 9: 289-291.
McAleese D.M., 1958. Studies on the basaltic soils of norther Ireland V. Cation-exchange capacities and mineralogy of the silt separates. Journal of Soil Science, 9: 81-88.
McBride M.B., 1976. Exchange and hydration properties of Cu+2 and mixed Na+-Ca2+ smectites. Soil Science Society of America Journal, 40: 452-456.
McBride M.B., Tyler L.D., and Hovde D.A., 1981. Cadmium adsorption by soils and uptake by plants as affected by soil chemical properties. Soil Science Society of America Journal, 45: 739-744.
Mckenzie F.T., Lantzi R.J., and Paterson V., 1979. Global trace metal cycles and predictions. Journal of International Association of Mathematical Geology, 11: 99-142.
McConaghy S., 1957. Studies on the basaltic soils of northern Ireland I. Cation-exchange properties. Journal of Soil Science, 8: 127-134.
McConaghy S. D.M., 1958. Studies on the basaltic soils of norther Ireland III. Exchangeable-cation contents of sand, silt, and clay separates. Journal of Soil Science, 9: 66-75.
McGrat S.P., and Lane P.W., 1989. An explanation for the apparent losse of of metals in a long-term field experiment with sewadge sludge. Environmental Pollution, 60: 235-256.
McLaren R.G., and Crawford D.V., 1973. Studies on soil copper I. The fractionation of copper in soils. Journal of Soil Science, 24: .
Mermut A.R., Jain J.C., Song Li R., Kerric R., Kozak L., and Jana S., 1996. Trace element concentrations of selected soils and fertilizers in Saskatchewan, Canada. Journal of Environmantal Quality, 25: 845-853
Miller M.M. and Ohlrogge A.J., 1958. Water-soluble chelating agents in organic materials: 1. Characterization of chelating agents and their reactions with trace metals in soils. Soil Science Society of America Procedings, 22: 225-228.
Miller W.P., and McFee W.W., 1983. Distribution of cadmium, zinc, copper, and lead in soils of industrial northwestern Indiana. Journal of Environmental Quality, 12: 29-33.
Mitchell B.D., Farmer V.C., and McHardy W.J., 1964. Amorphous inorganic materials in soils. Advances in Agronomy, 16: 327-383.
Mitchell P., and Barr D., 1995. The nature and significance of public exposure to arsenic: a rewiew of its relevance to South West England. Environmental Geochemistry and Health. 17: 57-82
Mogollon L. J., Bifano C., and Davies B. E., 1995. Distribution of metals in mechanical fractions of soils from a lake catchment in Venezuela. Environmental Geochemistry and Health, 17: 103-111.
Morrow Alison, Wiltshire Guy, and Hursthouse Andrew, 1997. An improved method for the simultaneous determination of Sb, As, Bi, Ge, Se, and Te by hydride generation ICP-AES: application to environmental samples. Atomic Spectroscopy, 18: 23-28.
Mortensen J.L., 1963. Complexing of metals by soil organic matter. Soil Science Society of America Proceedings, 27: 179-186.
Mosser C., Petit S., and Mestdagh M., 1992. ESR and IR evidence for chromium in kaolinites. Clay Minerals, 28: 353-364.
Murali V., and Aylmore L.A.G., 1981. Competitive adsorption: its importance in ionic transport in soils. Search, 12: 133-135.
Munza B., Deiana S., Maddau V.,Gessa C., and Seeber R., 1995. Stability constants of metal-humate complexes: titration data analized by bimodal gaussian distribution. Soil Science Society of America Journal, 59: 1570-1574.
Murali V., and Aylmore L.A.G., 1981. A convective-dispersive-adsorptive flow model for solute transport in soils: 1. Model description and simulations. Australian Journal of Soil Reserch, 19: 23-39.
Murali V., and Aylmore L.A.G., 1980. No-flow equilibration and adsorption dynamics during ionic transport in soils. Nature, 238: 467-469.
Murali V., and Aylmore L.A.G., 1983. Competitive adsorption during solute transport in soils: 3. A rewiew of experimental evidence of competitive adsorption and evaluation of simple competition models. Soil Science, 136: 279-290.
Murali V., and Aylmore L.A.G., 1983. Competitive adsorption during solute transport in soil: I. Mathematical models. Soil Science, 135: 143-150.
Murray J. W., and Dillard J.G., 1979. The oxidation of Co(II) adsorbed on manganese dioxide. Geochimica et Cosmochimica Acta, 43: 781-787.
Murray J. W., 1975. The interaction of metal ions at the manganese dioxide-solution interface. Geochimica et Cosmochimica Acta, 39: 505-519.
Murray R. S., 1979. “Teoria e problemi di probabilità e statistica”. Collana Shaum, ETAS Libri, Milano.
Nagarajah S., Posner A.M., and Quirk J.P., 1970. Competitive adsorption of phosphate with polygalacturonate and other organic anions on kaolinite and oxides surfaces. Nature, 228: 83-84
Negretti G., Sabatino B., 1983. “Corso di petrografia”, CISU, Roma.
Neihof R.A., and Loeb G., 1972. The surface change of particulate matter in seawater. Limnol. Oceanoggr., 17: 7-16.
Newman A.C.D., and Brown G., 1966. Chemical changes during the alteration of micas. Clay Minerals, 6: 297-310.
New Zeland Society of Soil Science, 1980. “Soil with Variable Charge”. Offset Publication, Palmerston North.
Nightingale E.R Jr, 1959. Phenomenological theory of ion solvatation. Effectivr radii of hydrated ion. Journal of Physicall Chemistry, 63: 1381-1387.
Nriagu J.O., and Pacyna J.M., 1988. Quantitative assesment of wordwide contamination of air, water and soils by trace metals. Nature, 1988: 333-134.
Nowland G.A., 1976. Concretionary manganese-iron oxides in stream and their usefulness as sample medium for geochemical prospecting. Journal of Geochemical Exploration, 6: 193-210.
Ochs M., Cosovic B., and Stumm W., 1994. Coordinative and Hydrofobic interaction of humic substances with hydrophilic Al203 and hydrophobic mercury surfaces. Geochimica et Cosmochimica Acta, 58: 639-650.
Olsen S.R., and Kemper W.D., 1968. Solute movement in the soil-root system. Advances in Agronomy, 20: 91-151.
O’Neill P., 1997. “Arsenic”. In: Alloway B.J. Ed, 1997. “Heavy Metals in soils”. Blackie Academic and Professional, London.
Outridge P.M., and Noller B.N., 1991. Accumulation of toxic trace metals by freshwater vascular plants. Review of Environmental Contamination and Toxicology, 115: 91-124.
Outridge P.M., and Scheuhammer A.M., 1993. Bioaccumulation and toxicology of chromium: implication for wildlife. Review of Environmental Contamination and Toxicology, 124: 79-110.
Page AL., Miller R.H., Keeeney D.R., Eds, 1982. “Methods for soil analysis, part II”, 2nd Edition, American Society of Agronomy, Madison, Wisconsin, USA.
Parfit R.L., and Russel J.D., 1977. Adsorption on hydrous oxides IV. Mechanism of adsorption of varius ions on goethite, Journal of Soil Science, 28: 297-305.
Parfitt R.L., 1978. Anion adsorption by soil and soil materials. Advances in Agronomy, 30: 1-50.
Pedro C., Jamagne M.m and Begon J.C., 1969. Mineral interactions and transformation in relation to pedogennesis during the Quaternary, Soil Science, 107: 462-469.
Peigneur P., Maes A., and Cremers A., 1975. Heterogeneity of charge density distribution in montmorillonite as inferred from cobalt adsorption. Clays and Clay Minerals, 23: 71-75.
Perret D., Leppard G.G., Muller M., Belzile N., De Vitre R.R., and Buffle J., 1991. Electron microscopy of acquatic colloids: non perturbing preparation of speciments in the field. Water Research, 25: 1333-1343.
Plotnikow V. I. and Usatova L. P., 1964. Coprecipitation of small amounts of arsenic with metal hydroxides. Zhirfrict Analitic Khimie, 19: 1183.
Poelstra P. and Frissel M.J., 1967. Migration of water and ions in indisturbate soil columns and its description by simulation models. In: “Isotope and radiation tecniques in soil physics and irrigation studies, proceedings symposium”, Istanbul, 12-16 June 1967, IEA, Vienna.
Quirk J.P., 1955. Significance of surface areas calculated from water vapor sorption isoterms by use of the B.E.T. equation. Soil Science, 80: 423-430.
Quraishi Yasmeen F., and Pandey G.S., 1995. Bronchial contamination with toxic metals in mineral-based industrial areas of India. Environmental Geochemistry and Health, 17: 25-28.
Ragan H.A., and Mast T.J., 1990. Cadmium inhalation and male reproductive toxicity. Review of Environmental Contamination and Toxicology, 114: 1-22.
Ramos L., Hernandez L.M., and Gonzalez M.J., 1994. Sequential Fractionation of copper, lead, cadmium and zinc in soils from or near Donana National Park. Journal of Environmenthal Quality. 23: 50-57.
Randhawa N.S., and Broadbent F.E., 1965. Soil organic matter complexes: 6. Stability constants of zinc-humic acid complexes at different pH values. Soil Science, 99: 362-365.
Reardon E.J., Allison G.B., and Fritz P., 1979. Seasonal chemical and isotopic variations of soil CO2 at Trout Creek, Ontario. Journal of Hydrology, 43: 355-371.
Reuter J.H., and Perdue E.M., 1977. Importance of heavy metal-organic matter interaction in natural watres. Geochimica et Cosmochimica Acta, 41: 325-334.
Rich C.I., 1964. Effect of cation size and pH on potassium exchange in Nason soil. Soil Science, 98: 100-106.
Rich C.I., and Blak W.R., 1964. Potassium exchange as affected by cation size, pH and mineral structure. Soil Science Society of America Proceedings, 97: 384-390.
Rochette E.A., Li G.C., and Fendorf S.E.. 1998. Stability of arsenate minerals in soil under biotically generated reducing conditions. Soil Science Society of America Journal, 62: 1530-1537.
Roy W.R., Hasset J.J., and Griffin R.A., 1986. Competitive coefficient for the adsorption of arsenate, molybdate, and phosphate mixtures by soils. Soil Science Society of America Journal, 50: 1176-1182.
Ruppert H, 1980. Fixation of metals on hydrous manganese and iron oxides phases in marine Mn-Fe-nodules and sediments. Chem. Erde, 39: 97-132.
Saar Robert A., and Weber James H., 1980. Lead(II) complexation by fulvic acid: how it differs from fulvic acid complexation of copper(II) and cadmium(II). Geochimica et Cosmochimica Acta, 44: 1381-1384.
Sabri Anmar W., Rasheed Kalid A., and Kassim Thaer I., 1993. Heavy metals in the water, suspended solids and sediment of the river tigris impoudment at samarra. Water Research, 6: 1099-1103.
Sadiq M., Zaida T.H., and Mian A.A., 1983. Environmental behavior of arsenic in soils: theoretical. Water Air and Soil Pollution, 20: 349-377.
Saeki Kazutoshi, Okazaki Masanori, and Matsumoto Satoshi, 1993. The chemical phase changes in heavy metals with drying and oxidation of the lake sediments. Water Research, 7: 1243-1251.
Sahl K., Doe B.R. and Wedepohl K.H., 1978. “Lead”. In Wedepohl K.H. Ed., 1978. “Handbook of Gechemistry”, Sprinnger-Verlag, Berlin.
Salomon W. and Förstner U., 1984. “Metals in the idrocycle”. Springer-Verlag, Berlin.
Santillan-Medrano J., and Jurinak J.J., 1975. The chemistry of lead and cadmium in soil: solid phase formation. Soil Science Society of America Journal, 39: 851-856.
Sawhney B.L., 1969. Regularity of interstratification as affected by charge density in layer silicate. Soil Science Society of America Procedeengs.
Sawhney B.L., 1970. Potassium and cesium ion selectivity in relation to clay mineral structure. Clays and Clay Mineral, 18: 47-52.
Sawhney B. L., 1972. Selective sorption and fixation of cation by clay minerals: a rewiew. Clays and Clay Mineral, 20: 93-100.
Schindler P.W., Furst B., Dick R., and Wolf P.U., 1976. Ligand properties of surface silanol groups. I. Surface complex formation with Fe+3, Cu+2, Cd+2, and Pb+2. Journal of Collid Interface Science, 55: 469-475.
Schindler P.W., Liechti P., and Westall J.C., 1987. Adsorption of copper, cadmium, and lead from aqueous solution to the kaolinite/water interface. Netherland Journal of Agricultural Science, 35: 219-230.
Schnitzer M, and Skinner S.I.M., 1966. Organo-metallic interactions in soils: 5. Stability constants of Cu2+, Fe2+, and Zn2+ fulvic acid complexes. Soil Science, 102: 361-365.
Schnitzer M., 1969. Reactions between fulvic acid, a soil humic compound and inorganic soil constituents. Soil Science Society of America Procedings, 33:75-81.
Schnitzer M., 1970. Characteristic of organic matter from podsol B horizons. Canadian Journal of Soil Science, 50: 199-204.
Schnitzer M. and Skinner M., 1964. Organo-metallic interaction in soils: 3. Properties of iron- and alluminium-organic matter complexes, prepared in the laboratory, and extracted from a soil. Soil Science, 98: 197-203.
Schnitzer M. and Skinner S.I.M., 1963. Organo-metallic interactions in soils: 1. Reactions between a number of metal ions and the organic matter of a Podsol Bh horizon. Soil Science, 96: 86-93.
Schnitzer M., and Hanson E.H., 1970. Organo-metallic interactions in soils: 8. An evaluation of mrthods for the determination of stability constants of metal-fulvic acid complexes. Soil Science, 109: 333-340.
Schnitzer M., and Hoffman I., 1967. Thermogravimetric analysis of the salts and metal complexes of a soil fulvic acid. Geochimica et Cosmochimica Acta, 31: 7-15.
Schnitzer M., and Levesque M., 1967. Organo-metallic interaction in soils: 6. Preparation and properties of fulvic acid-metal phosphates. Soil Science, 103: 183-190.
Schnitzer M., and Skinner S.I.M., 1965. Organo-metallic interaction in soils: 4. Carboxyl and phenolic hydroxyl groups in organic matter and metal retention. Soil Science, 99: 278-284.
Schnitzer M., and Skinner S.I.M., 1967. Organo-metallic interactions in soils: 7. Stability constants of Pb2+, Ni2+, Mn2+, Co2+, Ca2+, and Mg2+ fulvic acid complexes. Soil Science, 103: 247-252.
Schnitzer M., 1991. Soil organic matter - The next 75 years. Soil Science, 151: 41-58.
Schnor J.L., 1996. “Environmental Modeling, Fate and Transport of Pollutants in Water, Air, and Soil”. Environmental Science and Tecnology Series, Wiley and Soons, New York.
Schofield R.K., 1949. Effect of pH on electric charges carried by clay particles. Journal of Soil Science, 1: 1-8.
Schoof Rosalind A., Butcher Mattew K., Sellstone Christopher, Ball Wayne R., Fricke James R., Keller Vincent, and Keehn Barbara, 1995. An assesment of lead absorption from soil affected by smelter emissions. Environmental Geochemistry and Health, 17: 189-199.
Schulte A., and Beese F., 1994. Isoterms of cadmium sorption density. Journal of Environment Quality, 23: 712-718.
Schulthess C.P., and Huang C.P., 1990. Adsorption of heavy metals by silicon and alluminium oxides surfaces on clay minerals. Soil Science Society of America Journal, 54: 679-688.
Schulthess C.P., and Sparks D.L., 1986. Backtitration technique for proton isotherm modeling of oxide surfaces. Soil Science Society American Journal, 50: 1406-1411.
Schwarzen G. and Sillen G. L., 1958. “Stability Constants of Metal-Ion Complexes With Solubility Products of Inorganic Substances”. The Chemical Society, London.
Schwarzman D.W., and Walk T., 1989. Biotic enhancement of the weathering and the habitability of the hearth. Nature, 340: 457-460.
Sequi P. Ed, 1989. “Chimica del suolo”. Patron Editore, Bologna.
Shacklette H.T. and Boerngen J.B., 1984. Element concentrations in soils and other surficial materials of the conterminous United States. U.S. Geological Survey Prof. Paper.
Shainberg I., and Kemper W.D., 1966. Hydration status of adsorbed cations. Soil Sience Society of America Proceedings, 30: 707-713.
Shainberg I., and Kemper W.D., 1967. Ion exchange equilibria on montmorillonite. Soil Science, 103: 4-9.
Shan Xiao-Quan, and Chen Bin, 1993. Evaluation of sequential extraction for speciation of trace metals in model soil containing natural minerals and humic acid. Analitical Chemistry, 65: 802-807.
Sharmasarkar Shankar, and Vance George F., 1995. Fractional partitioning for assessing solid-phase speciation and geochemichal transformations of soil selenium. Soil Science, 160: 43-55.
Shas Matigod V., Gibali A.S., and Page A.L., 1979. Effect of ionic srength and ion pair formation on the adsorption of nickel by kaolinite. Clays and Clay Minerals, 27: 411-416.
Sheindorf C.H., Rebhun M., and Sheintuch M., 1981. A Freundlich type multicomponent isotherm. Journal of Colloid Interface Science, 79: 136-142.
Sheppard Marsha I., and Thibault Denis H., 1992. Desorption and extraction of selected heavy metals from soils. Soil Science Society of America Journal, 56: 415-423.
Sheppard S.C., 1995. A model to predict concentration enrichment of contaminants on soil adhering plants and skin. Environmental Geochemistry and Health, 17: 13-20.
Sheppard S.C., and Eveden W.G., 1990. Characteristics of plant concentration ratios assessed in a 64-site field survey of 23 elements. Journal of Environmenthal Radioactivity, 11: 15-36.
Sheppard S.C., Eveden W.G., and Schwartz W.J., 1994. Ingested soil: bioavailability of sorbed lead, cadmium, cesium, iodine, and mercury. Journal of Environmenthal Quality, 24: 498-505.
Sheppard S.C., Gaudet C., Sheppard M.I., Cureton P.M., and Wong M.P., 1992. The development of assesment and remediation guidelines for contaminated soils, a rewiew of the science. Canadian Journal of Soil Science, 72: 359-394.
Shindler P.W., Liecti P., and Westall J.C., 1987. Adsorption of copper, cadmium and lead from aqueous solution to the kaolinite/water interface. Neth. J. Agric. Sci., 35: 219-230.
Shirado T., Vergara I., Schlasha E.B., and Pratt P.F., 1986. Evidence for movement of heavy metals in soils irrigated with untreated wastewater. Journal of Environmental Quality, 15: 9-12.
Shirata H., Elias R.W., and Patterson C.C., 1980. Chronological variations in concentrations and isotopic compositions of anthropogenic atmospheric lead in sediments of a remote subalpine pond. Geochimica et Cosmochimica Acta, 44: 149-162.
Shulte Andreas, and Beese Fritz. 1994. Isotherms of cadmium sorption density. Journal of Environmenthal Quality, 23: 712-718.
Shulthess C.P., and Sparks D.L., 1989. Competitive ion exchange behaviour on oxides. Soil Science Society of America Journal. 53: 366-373.
Shulthess C.P., and Sparks D.L., 1986. Backtitration tecnique for proton isoterm modeling of oxide surfaces. Soil Science Society of America Journal, 50: 1406-1411.
Shulthess C.P., and Sparks D.L., 1988. A critical assesment of surface adsorption models. Soil Science Society of America Journal, 52: 92-97.
Shulthess C.P., and Sparks D.L., 1989. Competitive ion exchange behavior on oxides. Soil Science Society of America Journal, 53: 366-373.
Shulthess C.P., and Sparks D.L., 1988. A critical assesment of surface adsorption models. Soil Science Society American Journal, 52: 92-97.
Shuman L.M., 1979. Zinc, Manganese, and copper in soil fractions. Soil Science, 127: 10-17.
Shuman L.M., 1981. Seaparating soil iron- and manganese-oxides fractions for microelements analysis. Soil Science Society of America Journal, 46: 1099-1102.
Shuman L.M., 1985. Fractionation method for soil microelements. Soil Science. 140: 11-22.
Sibbesen E., and Andersen C.E.. 1985. Soil movement in long term field experiments as result of cultivation: II. How to estimate the two-dimensional movement of substances accumulating in the soil. Exp. Agric., 21: 109-117.
Sibbesen E., Andersen C.E., Andersen S., and Flested-Jensen M., 1985. Soil movement in a long-term field experiments as a result of cultivations: I. A model for approximating soil movement in one horizontal dimension by repeted tillage, Exp. Agric., 21: 101-107.
Sidle R.C., and Kardos L.T., 1977. Transport of heavy metals in a sludge-treated forested area. Journal of Environmental Quality, 6: 431-437.
Sidle R.C., Kardos L.T., and van Genucten M. Th., 1977. Heavy metal transport in a sludge treated soil. Journal of Environmental Quality, 6: 438-443.
Sidle R.C., Sopper W.E., 1976. Cadmium distribution in forest ecosystem irrigated with treated municipal waste water and sludge. Journal of Environmental Quality, 5: 419-422.
Sienco M.J. e Plane R.A., 1980. “Chimica. Principi e proprietà”. Piccin Editore, Padova.
Sillampää M., 1982. “Micronutrients and the nutrient status of soils: a global study”. FAO Soil Bulletin n°48, Roma.
Silviera D.J., and Sommers L.E., 1977. Extractability of copper, zinc, cadmium, and lead in soils incubated with sewage sludge, Journal of Environmental Quality, 6: 47-52.
Sims J.L., Duangpatra Piya, Ellis J.H., and Phillips R.E., 1979. Distribution of available manganese in kentucky soils. Soil Science, 127: 270-274.
Snitzer M. and Kodama H., 1966. Montmorillonite: effect of pH on its adsorption of a soil humic compound, Science, 153: 70-71.
Snitzer M., 1991. “Soil organic matter-the next 75 years. Soil Science. 151: 41-58.
Società Italiana della Scienza del Suolo, 1985. Metodi normalizzati di analisi del suolo, Edagricole, Bologna.
Sommers L.E., and Lindsay W.L., 1979. Effect of pH on predicted heavy metal-chelate equilibria in soils. Soil Science Society of America Journal, 43: 39-47.
Soon Y.K.. 1994. Changes in forms of soil zinc after 23 years of cropping following clearing of a boreal forest. Canadian Journal of Soil Science, 74: 179-183.
Sposito G., 1989. “The chemistry of soils”. Oxford University Press, Oxford.
Sposito G., and Holtzclaw K.M., Johnston C.T., and LeVesque-Madore C.S., 1981. Thermodinamics of sodium-copper exchange on Wyoming bentonite at 298K. Soil Science Society of American Journal, 45: 1079-1084.
Sposito G., Bingham F.T., Yadav, S.S., and Inouye Carol A.. 1982. Trace metal complexation by fulvic acid extracted from sewage sludge: II. Development of chemical models. Soil Science Society of America Journal. 46: 51-56.
Sposito G., Lund L.J., and Chang A.C., 1980. Trace metal chemistry in arid-zone field soils amended with sewage sludge: I. Fractionation of Ni, Cu, Zn, Cd, and Pb in solid phases. Soil Science Society of America Journal, 46: 260-264.
Stigliani W.M., 1992. Overwiew of the chemical time bomb problem in Europe. In: Muelen G.R.B., Stiglinai W.M., Salomons W., Bridges E.M., and Imenson A.C., Eds, 1993. Chemical time bomb. Proceedinngs of the european state-of-theart conference of the delayed effects of the chemicals in soils and sediments. Veldhoven, The Netjherland, 2-5 september 1992.
Stevenson F.J., 1976. Stability constant of Cu+2,Pb+2, and Cd+2 complexes with humic acids. Soil Science Society of America Journal, 40: 665-672.
Stevenson F.J., 1977. Nature of divalent transition metal complexes of humic acids as revelated by a midified potentiometric titration method. Soil Science, 173: 10-17.
Stevenson F.J. Krastanov S.A., and Ardakani M.S., 1973. Formation constant of Cu2+ complexes with humic and fulvic acids. Geoderma, 9: 129-141.
Stevenson F.J., and Chen Y., 1991. Stability constants of copper(II)-humate complexes determined by modified potentiometric titration. Soil Science Society of America Journal, 55: 1586-1591.
Stevenson F.J., and Goh K.M., 1971. Infrared spectra of humic acids and related substances. Geochimica et Cosmochimica Acta, 35: 471-483.
Stevenson F.J., and Goh K.M., 1974. Infrared spectra of humic acids: elimination of interference due hygroscopic moisture and structural changes accompanying heating with KBr. Soil Science, 117: 34-41.
Stevenson F.J., and Welch L.F., 1979. Migration of applied lead in a field soil. Environmental Science and Technology, 13: 1255-1259.
Stevenson F.J., Fitch Alan, and Brar M.S., 1993. Stability constants of Cu(II)-humate complexes: comparision of select models. Soil Science, 155: 77-91.
Stumm W., Hohl H., and Dalang F., 1976. Interaction of metals ions with hydrous oxide surfaces. Croatica Chemica Acta, 48: 491-504.
Stumm W., Kummert R., and Sigg L., 1980. Aligand exchange model for the adsorption of inorganic and organic ligands ad hydrous oxide interfaces. Croatica Chemica Acta, 53: 291-312.
Stumm W., and Morgan J.J., 1996. “Aquatic Chemistry”. Environmental Science and Tecnology Series, Wiley and Soons, New York.
Swallow K.C., Hume D.N., and Morel F.M.M., 1980. Sorption of cupper and lead by hydrous ferric oxides. Environmental Science and Technology, 14: 1326-1331.
Takamatsu T., and Yoshida T., 1978. Determination of stability constants of metal-humic acid complexes by potentiometric titration and ion-selective electrodes. Soil Science, 125: 377-386.
Tamaki S., and Frankenberg W.T. Jr, 1992. Environmental biogeochemistry of arsenic. Review of Environmental Contamination and Toxicology, 124: 79-110.
Tan K.H., King L.D., and Morris H.D., 1971. Complex reaction of zinc with organic matter extracted from sewadge sludge. Soil Science Society of America Proceedings, 35: 748-751.
Tan K.H., Leonard R.A., Bertrand A.R., and Wilkinson S.R., 1971. The metal complexing capacity and the nature of the chelating ligands of water extractc of poultry litter. Soil Science Society of America Proceedings. 35: 265-269.
Teraoaka H. and Kobayashi J., 1980. Cocentration of 21 Metals inthe Suspended Solid Collected from the Principal 166 Rivers and 3 Lakes in Japan. Geochemistry Journal, 14: 203-226.
Terzaghi P., 1967. “Geotecnica”. UTET, Torino.
Tessier A., Campbell P.G.C., and Bisson M., 1979. Sequential extraction procedure for the speciation of particulate trace metals. Analitical Chemistry, 51: 844-851.
Tessier A., Fortin D., Belzile N., DeVitre R.R., and Leppard G.G., 1996. Metal sorption to diagenetic iron and manganese oxydroxides and associated organic matter: narrowing the gap between field and laboratory measurements. Geochimica et Cosmochimica Acta, 60: 387-404.
Testini C. e Gessa C., 1989. “Le fasi solide”. In: Sequi P. Ed., 1989. “Chimica del suolo”, Patron Editore, Bologna.
Thorez J., 1976. “Pratical identification of clay minerals” Lelotte éd Disan.
Tipping E., 1981. The adsorption of acquatic humic substances by iron oxides. Geochimica et Cosmochimica Acta, 45: 191-199.
Tipping E., and Woof C., 1990. Humic substances in acid organic soils: modelling their release to the soil solution in terms of humic charge. Journal of Soil Science, 41: 573-586.
Travis C.C., and Hester S.T., 1991. Global chemical pollution. Environmental Science and Technology, 25: 815- .
Tucker B.M., 1974. Displacement of ammonium ions for cation exchange capacity measurements. Journal of Soil Science, 25: 333-337.
Turekian K.K., 1977. The fate of metals in the oceans. Geochimica et Cosmochimica Acta, 41: 1139-1144.
Tyler L.D., and McBride Murray B., 1981. Mobility and extractability of cadmium, cooper, nickel, and zinc in organic and mineral soil colums. Soil Science, 134: 198-205.
van Dijk H, 1971. Cation binding of humic acids. Geoderma, 5: 53-67.
Violante P., 1989 a. “I fattori di formazione del suolo”. In: Sequi P., 1989. “Chimica del suolo”, Patron Editore, Bologna.
Violante P., 1989 b. “I processi di alterazione”. In: ”. In: Sequi P., 1989. “Chimica del suolo”, Patron Editore, Bologna.
Vinogradov A.P., 1959. “The geochemistry of rare and dispersed chemicals elements in soils”, 2nd Edition. Chapman and Hall, London.
Wagner G.H., and Stevenson F.J., 1965. Structural arrangement of funtional groups in soil humic acids as revelated by infrared analysis. Soil Sience Society of America Proceedings, 29: 43-48.
Wahlen M., and Thompson R.C., 1980. Pollution records from sediments of tree lakes in New York State. Geochimica et Cosmochimica Acta, 44: 333-339.
Wedepohl K.H. Ed, 1969. “Handbook of Geochemistry”, Springer-Verlag, Berlin.
Whitfield M., Turner D.R., 1982. Ultimate removal mechanisms of elements from the ocean - a comment. Geochimica et Cosmochimica Acta, 46:1989-1992.
Whitmore F., Maestri
B., Mabey W., Holt B., and Gould C, 1979. “Water-Related Environmental Fate
of 129 Priority Pollutants. Volume I: Introduction and Technical Background,
Metals and Inorganics, Pesticides and PCBs”. EPA-440/4-79-029a, EPA contracts
68-01-3852 and 68-01-3867, Office of Water Planning and Standards, U.S.
Environmenntal Protection Agency, Wascington D.C. Citato in Bodec I. et al., 1988.
Williams D.E., Vlamis J., Pukite A.H., and Corey J.E., 1980. Trace elements accumulation, movement, and distribution in the soil profile from massive applications of seavadge sludge. Soil Science, 129: 119-132.
Williams D.E., Vlamis J., Pukite A.H., and Corey J.E., 1985. Metal movement in sludge-treated soils after six years of sludge addition: II. Nickel, iron, manganese, cromium, and mercury. Soil Science, 140: 120-125.
Williams D.E., Vlamis J., Pukite A.H., and Corey J.E., 1987. Metal movement in sludge-amended soils: a nine-year study. Soil Science, 143: 124-131.
Williams D.J.A., and Williams K.P., 1978. Electrophoresis and zeta potential of kaolinite. Journal of Colloid Interface Science, 65: 79-87.
Wilson M.J., 1987. “Handbook of determinative methods in clay mineralogy”. Blakie and Soons, London.
Wood J.C., Moschopedis S.E., and den Hertog W., 1961. Study in humic acid chemistry. II: humic anhydrides. Fuel, 40: 491-502.
Wright J.R., Levick R., and Atkinson H.J., 1955. Trace element distribution in virgin profiles representing four Great Soil Groups. Soil Science Society of America Journal, 19: 340-345.
Yatsimirskii K. B. and Vasil’ov V.P., 1960. “Instability Constants of Complex Compuonds”, Pergamon Press, London.
Yasonuri Mahara, 1993. Storage and migration of fallout strontium-90 and cesium-137 for over 40 years in the surface soil of nagasaki. Journal of Envirmenthal Quality, 22: 722-730.
Yingming L., and Corey R.B., 1993. Redistribution of sludge-borne cadmium, copper, and zinc in a cultivated plot. Journal of Environmental Quality, 22: 1-8.
Yuan-Hui Li, 1982. Interelement relationship in abyssal pacific ferromanganese nodules and associated pelagic sediments. Geochimica et Cosmochimica Acta, 46:1053-1060.
Yu Shane Y., Bailey G.W., and Xianchan J., 1996. Application of a lumped, nonlinear kinetics model to metal sorption on humic substances. Journal of Environmantal Quality, 25: 552-561.
Yu Shane Y., Bailey George W., and Xianchan Jin, 1996. Application of a lumped, nonlinear kinetics model to metal sorption on humic substances. Journal of Environmantal Quality, 25: 552-561.
Zach Reto, and Sheppard Steve C., 1991. Food-chain and dose model, CALDOS, for assessing Canada's nuclear fuel waste managment concept. Health Physics, 60: 643-656.
Zachara J.M., Ainsworth C.C., Cowan C.E., Resch C.T., 1989. Adsorption of chromate by subsurface soil horizons. Soil Science Society of America Journal, 53: 418-428.
Ziper C., Komarneni S., and Baker D.E., 1988. Specific cadmium sorption in relation to the crystal chemistry of clay minerals. Soil Science Society of America Journal, 52: 49-53.
Zunino H., Peirano P., Aguilera M, and Schalscha E.B., 1975. Measurement of metal complexing ability of polifuntional macromolecules: a discussion of the relationship between the metal complexing properties of the extracted soil organic matter and soil genesis and plant nutrition. Soil Science, 119: 210-216.
Zunino H., Peirano P., Agulera M., and Escobar I., 1972. Determination of maximum complexing ability of water-soluble complexants. Soil Science, 114: 414-416.